2022/10/17 help更新
2023/01/05 論文引用、07/26 追記
微生物リファレンスゲノムの増加により、メタゲノム解析の精度は向上したが、分類学的プロファイラーのインデックス作成効率、データベースサイズ、実行時間に対する要件は高くなってきている。また、多くのプロファイラーは、主に細菌、古細菌、真菌の集団に焦点を当て、ウイルスの集団にはあまり注意を払っていない。本発表では、k-merを用いたメタゲノムプロファイリングツールKMCPを紹介する。KMCPは、リファレンスゲノムをチャンクに分割し、k-merにゲノム上の位置を導入する。シミュレーションデータと実データを用いたベンチマーク結果から、KMCPはメタゲノムショットガン配列データから古細菌、バクテリア、ウイルス集団を正確に分類学的にプロファイリングできるだけでなく、デプスの浅い感染性臨床サンプルに対して確信を持って病原体を検出できることが実証された。KMCPはGoで実装され、MITのもとオープンソースソフトウェアとして、https://github.com/shenwei356/kmcp で公開されている。
2023/07
KMCP v0.9.3 fixed two tiny bugs and improved the log information.
— Wei Shen 沈伟 (@shenwei356) July 16, 2023
And two tutorials were added according to users' needs:
1. Detecting specific pathogens from sequencing data.
2. Detecting contaminated sequences in a bacterial assembly.https://t.co/inrPQS5w8V
🎉KMCP is out in Bioinformatics. In the paper we present a novel k-mer-based metagenomic profiling tool that combines k-mer similarity and genome coverage information to increase the profiling accruacy.https://t.co/uOSO6GNFXI
— Wei Shen 沈伟 (@shenwei356) December 30, 2022
Can't wait to share the KMCP preprint with you, hope it's not too late. Thanks for reading and any feedback is welcome and appreciated. https://t.co/mVZJ6k1jCi https://t.co/GPIxPBfMln
— Wei Shen 沈伟 (@shenwei356) March 9, 2022
Database (Taxonomy fileも有り)
https://bioinf.shenwei.me/kmcp/database/
KMCPの特徴(レポジトリより)
- 正確なメタゲノムプロファイリング。KMCPは、リファレンスゲノムを10チャンクまたは5チャンクに分割し、そのチャンクに高速k-merマッチングによりリードをマッピングする、新しいメタゲノムプロファイリング戦略を採用しています(疑似マッピングと呼ぶことにします)。シミュレーションデータと実データを用いたベンチマークの結果、KMCPはメタゲノムショットガン配列データから古細菌、バクテリア、ウイルス集団の正確な分類学的プロファイリングを可能にするだけでなく、低深度の感染性臨床サンプルにおいて確信を持って病原体を検出できることがわかった(ベンチマークを確認して下さい)。
- 大規模ゲノムデータに対する高速配列検索。KMCPは、BIGSIやCOBSのように大規模なゲノムデータに対する高速な配列検索に使用することができます。COBS(Compact Bit-Sliced Signature index)アルゴリズムを再実装・改良し、インデックスサイズを小さくし、検索速度を大幅に向上させました(COBSの4倍〜10倍)(チュートリアルとベンチマークを参照)。
- ゲノム類似性の高速推定。KMCPは、既知のリファレンスゲノムに対するゲノムアセンブリの類似性を高速に推定するためにも使用できます。
- データベースの構築は簡単で高速。GTDB-r202の47894ゲノムに対して、40CPUスレッドとソリッドディスクドライブ搭載のサーバーで約25分。
- 高速な検索。利用可能なすべてのCPUコアを利用するように自動的にスケールします。検索時間は参照ゲノム数に線形に比例します。
- スケーラブルな検索。複数のデータベースに対する検索結果を高速にマージすることができます。これにより、多くの利点が得られます。参照ゲノムの追加に伴うデータベースの再構築が不要になります。HPCクラスタでは、各計算ノードが参照ゲノムの一部で構築されたデータベースをホストすることにより、検索をリニアに高速化することができます。メインメモリに制限のあるコンピュータでも、小さなデータベースを構築することにより、検索をサポートすることができます。
- 正確な分類学的プロファイリング。K-merを用いた分類学的プロファイラーの中には、誤検出率が高いものがありますが、KMCPは複数の戦略を採用することで特異性を高め、同時に高い感度を維持しています。古細菌、バクテリアに加え、ウイルス/ファージでも良好な結果が得られました。KMCPは、感染性病原体の検出にも自信を持っています。
- 6つのモードをプリセットし、複数のシナリオに対応。
- CAMIおよびMetaPhlAnプロファイリングフォーマットに対応。
インストール
ubuntu18でテストした。
#conda (link)2023年7月現在v0.9.3
mamba create -n kmcp -y
conda activate kmcp
mamba install -c bioconda kmcp -y
> kmcp --help #v0.9.0
$ kmcp --help
Program: kmcp (K-mer-based Metagenomic Classification and Profiling)
Version: v0.9.0
Documents: https://bioinf.shenwei.me/kmcp
Source code: https://github.com/shenwei356/kmcp
KMCP is a tool for metagenomic classification and profiling.
KMCP can also be used for:
1. Fast sequence search against large scales of genomic datasets
as BIGSI and COBS do.
2. Fast assembly/genome similarity estimation as Mash and sourmash do,
by utilizing Minimizer, FracMinHash (Scaled MinHash), or Closed Syncmers.
Usage:
kmcp [command]
Available Commands:
autocompletion Generate shell autocompletion script
compute Generate k-mers (sketches) from FASTA/Q sequences
index Construct database from k-mer files
merge Merge search results from multiple databases
profile Generate taxonomic profile from search results
search Search sequences against a database
utils Some utilities
version Print version information and check for update
Flags:
-h, --help help for kmcp
-i, --infile-list string ► File of input files list (one file per
line). If given, they are
appended to files from CLI arguments.
--log string ► Log file.
-q, --quiet ► Do not print any verbose information.
But you can write them to file
with --log.
-j, --threads int ► Number of CPUs cores to use. (default 128)
Use "kmcp [command] --help" for more information about a command.
実行方法
1、2は公開されているD.B(link)を使う場合は不要。
1、k-merの計算。
kmcp compute -k 21 --split-number 10 --split-overlap 100 \
--in-dir genomes/ --out-dir genomes-k21-n10
genomes-k21-n10/

2、k-merのインデックスを作成。
kmcp index --false-positive-rate 0.1 --num-hash 1 \
--in-dir genomes-k21-n10/ --out-dir genomes.kmcp
rm -rf genomes-k21-n10/
genomes.kmcp/
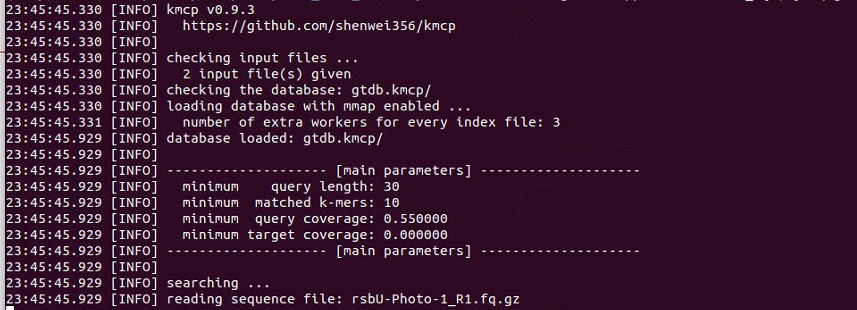
3、検索。fastqを指定する。データベースのロードに時間がかかる。読み込みの早いSSDにDBを配置した方がカウント開始までの時間が早くなる。スレッド数40指定。
kmcp search --db-dir genomes.kmcp/ input_R{1,2}.fastq.gz --out-file search.kmcp@db1.kmcp.tsv.gz
#公開されているGTDB D.B(link)を使うなら、ダウンロードしたターボールを解凍してそのディレクトエリを指定
kmcp search --db-dir gtdb.kmcp/ input_R1.fastq.gz input_R2.fastq.gz --out-file search_result.tsv.gz -j 40
#もしくはgtdb.kmcp/name.mapも指定してtaxonomyをアサインする(4のprofilingで使うとエラーを起こした)
kmcp search --db-dir gtdb.kmcp/ input_R1.fastq.gz input_R2.fastq.gz --out-file search_result.tsv.gz -j 40 -N gtdb.kmcp/name.map
4、プロファイリング
taxdumpファイルが必要。linkから taxdump.tar.gzをダウンロードして解凍した。このディレクトリのパスを--taxdumpで指定する。--taxid-mapでD.Bを指定する。ここではgtdb.kmcp/となる。また、3の出力(.tsv.gz)を指定する。
kmcp profile --taxid-map gtdb.kmcp/taxid.map --taxdump taxdump/taxid.map search_result.tsv.gz \
--out-prefix kmcp.profile \
-metaphlan-report metaphlan.profile \
--cami-report cami.profile \
--sample-id "0" \
--binning-result binning.gz
- -X, --taxdump Directory of NCBI taxonomy dump files: names.dmp, nodes.dmp, optional with merged.dmp and delnodes.dmp.
- -T, --taxid-map Tabular two-column file(s) mapping reference IDs to TaxIds.
複数D.Bを使っていてtaxid.mapファイルが複数ある場合、それらを連結して大きなtaxid.mapを作成し、それを指定する。KMCPの出力は各taxonを全分類ランクで計算するのではなく、対象リファレンスに対してのみアバンダンスを計算する。そのため、CAMIまたはMetaPhlAn形式でも出力することが推奨されている。
引用
KMCP: accurate metagenomic profiling of both prokaryotic and viral populations by pseudo-mapping
Wei Shen, Hongyan Xiang, Tianquan Huang, Hui Tang, Mingli Peng, Dachuan Cai, Peng Hu, Hong Ren
bioRxiv, Posted March 08, 2022
KMCP: accurate metagenomic profiling of both prokaryotic and viral populations by pseudo-mapping
Wei Shen, Hongyan Xiang, Tianquan Huang, Hui Tang, Mingli Peng, Dachuan Cai, Peng Hu, Hong Ren
Bioinformatics, Published: 29 December 2022
関連