レポジトリより
BUSCOMPはBUSCOの非決定論的限界を克服するために設計されている。アセンブリから完全なBUSCOの冗長でない最大集合をコンパイルし、この集合を用いて同じゲノムの異なるアセンブリ間の完全性の "真の "比較を予測可能な挙動で提供する。各BUSCO遺伝子について、BUSCOMPはfull_table_*.tsv結果テーブルとヒット配列のsingle_copy_busco_sequences/ディレクトリを使用して、利用可能な配列から最良の "Single Complete "配列を抽出する。BUSCOMPは全てのアセンブリ中のヒットをScoreでランク付けし、上位のヒットを保持する。タイは長さで解決され、最も長い配列が保持される。ScoreとLengthが同点の場合は、任意のエントリーを勝者として保持する。Single cmpleteヒットは、たとえスコアが低くてもDuplicateヒットより優先される。なぜならSingleヒットだけがBUSCOによってsingle_copy_busco_sequences/ディレクトリに配列が保存されているからである。この予測遺伝子配列セットが "BUSCOMPSeq "遺伝子セットとなる。
BUSCOMPはminimap2を使用して、BUSCOSeqで予測されたCDS配列を、オリジナルのBUSCOコンパイルに含まれていないものも含むゲノム/トランスクリプトームアセンブリにマッピングする。このように、種特異的なBUSCO配列のコンパイルされたセットは、新しいゲノムアセンブリの完全性を素早く評価するために使用することもできる。ヒットはパーセンテージカバレッジスタットに変換され、カバレッジと同一性に基づいてBUSCO遺伝子を再分類するために使用される。BUSCOMPレーティングはオリジナルのBUSCOレーティングを模倣するように設計されているが、定義が異なる。さらに、低品質ヒットのクラスとして "Partial "と "Ghost "が追加された。
Complete: 単一のコンティグ/スキャフォールドで95%以上のカバレッジ。(注:精度/同一性は考慮されない)
Duplicated: 2つ以上のコンティグ/スキャフォールドで95%以上のカバレッジ。
Fragmented:95%以上の複合カバレッジだが、単一のコンティグ/スキャフォールドにはない。
Partial:40~95%の複合カバレッジ。
Ghost: ローカルカットオフを満たすが、複合カバレッジが40%未満のヒット。
Missing: ローカルカットオフを満たすヒットがない。
注:ここで重複遺伝子とは、2つの異なるコンティグ/スキャフォールドでヒットした遺伝子を指す。BUSCOMPは詳細な完全性評価よりも、アセンブリのcoding potential を特定することに重点を置いている。同じコンティグ/スキャフォールド上の重複遺伝子はCompleteとマークされる。
個々のアセンブリ統計に加え、BUSCOとBUSCOMPの評価は、様々な出力を持つユーザー定義のアセンブリーグループを横断して集計され、異なるアセンブリがどのように相互補完しているかを知ることができる。また、評価は、与えられたゲノムサイズ=Xに基づく従来のゲノムアセンブリ統計(NG50とLG50)と組み合わされ、「ベスト」アセンブリを特定するのに役立つ。設定の詳細、主要な結果、表、プロットは、Rmarkdownを使用してHTMLレポートに出力される。
Slides
https://f1000research.com/slides/8-995
インストール
python3.12の環境を作ってテストした。Rのバージョンは4.4.3(HTML outputにはRとRmarkdownなどが必要)
#依存
mamba install --c bioconda minimap2 -y
mamba install conda-forge::pandoc -y
#Rmarkdown
> R
> install.packages("rmarkdown")
> install.packages("knitr")
> install.packages("ggrepel")
git clone https://github.com/slimsuite/buscomp.git
cd buscomp
#対話モードでの実行
python3 buscomp/code/buscomp.py
実行方法
1,まずBUSCOを各ゲノムに対して実行する。ここではbacteria_odb12 を指定
for GENOME in ./*.fna; do
GENBASE=$( basename ${GENOME/.fna/} )
busco -m genome -o run_$GENBASE -i $GENOME -l bacteria_odb12 --cpu 12
done
出力ディレクトリの1つ(例)
各busco出力ディレクトリにあるshort_summary.txtとfull_table.tsv(サブディレクトリにある)が必要。どちらもrun_bacteria_odb12下にあるのが認識される。
2,buscomp.pyの実行。log=debug -i -1をつけないと対話モードになるので注意。
git clone https://github.com/slimsuite/buscomp.git
python3 buscomp/code/buscomp.py indir=$PWD basefile=busco_comp_short busco=short combine=T summarise=T log=debug -i -1
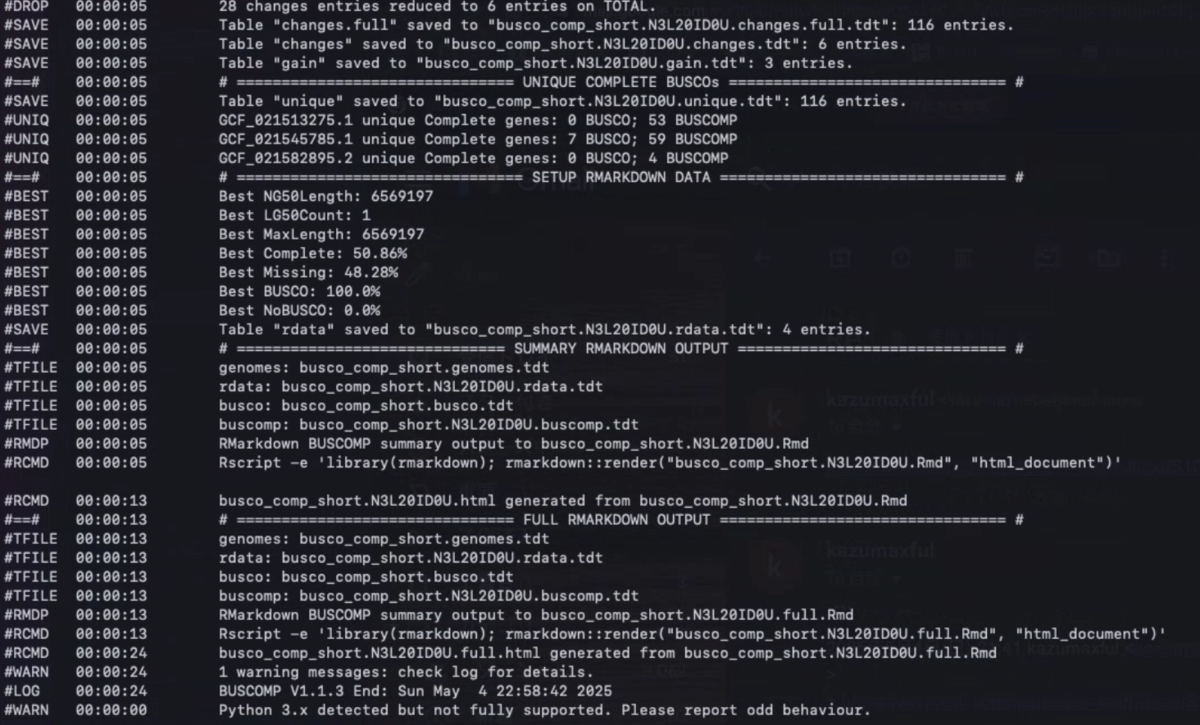
HTMLレポート例



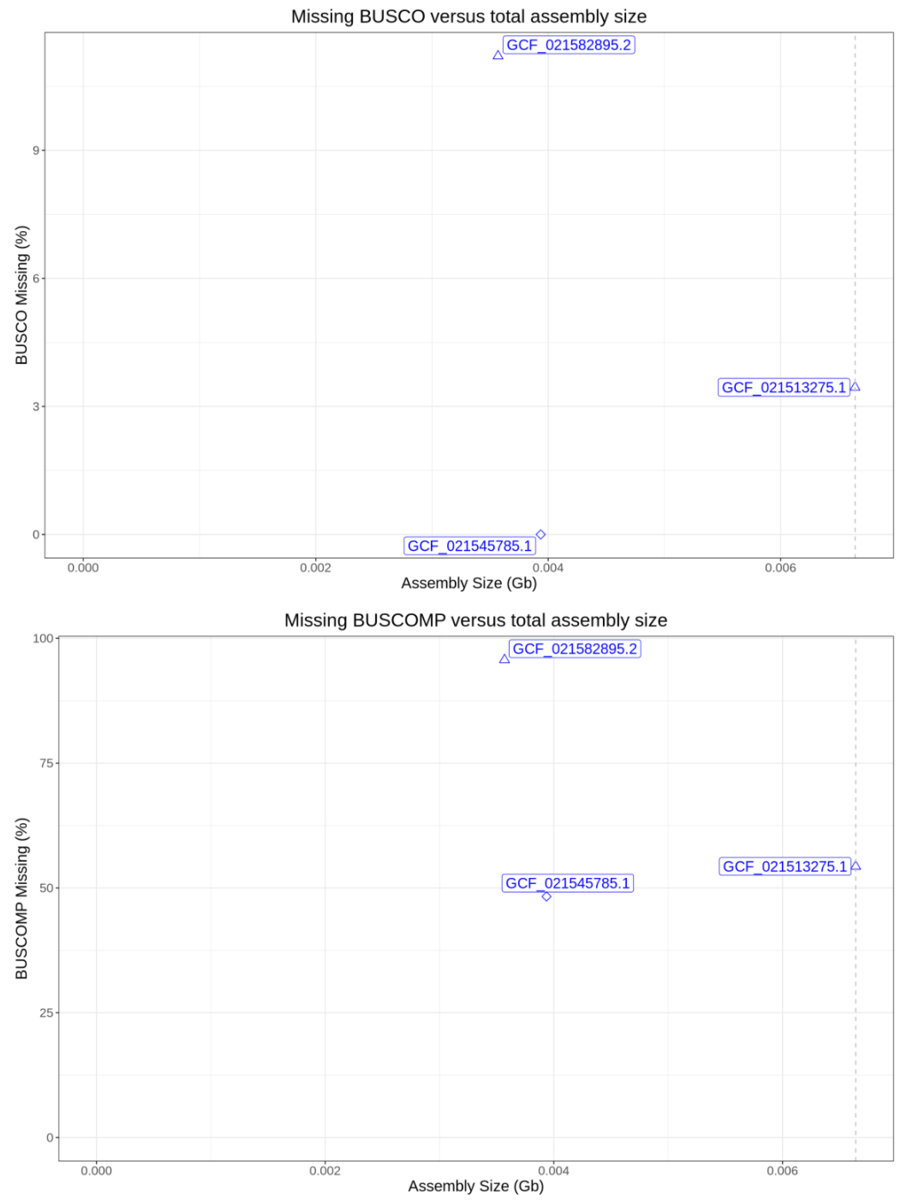


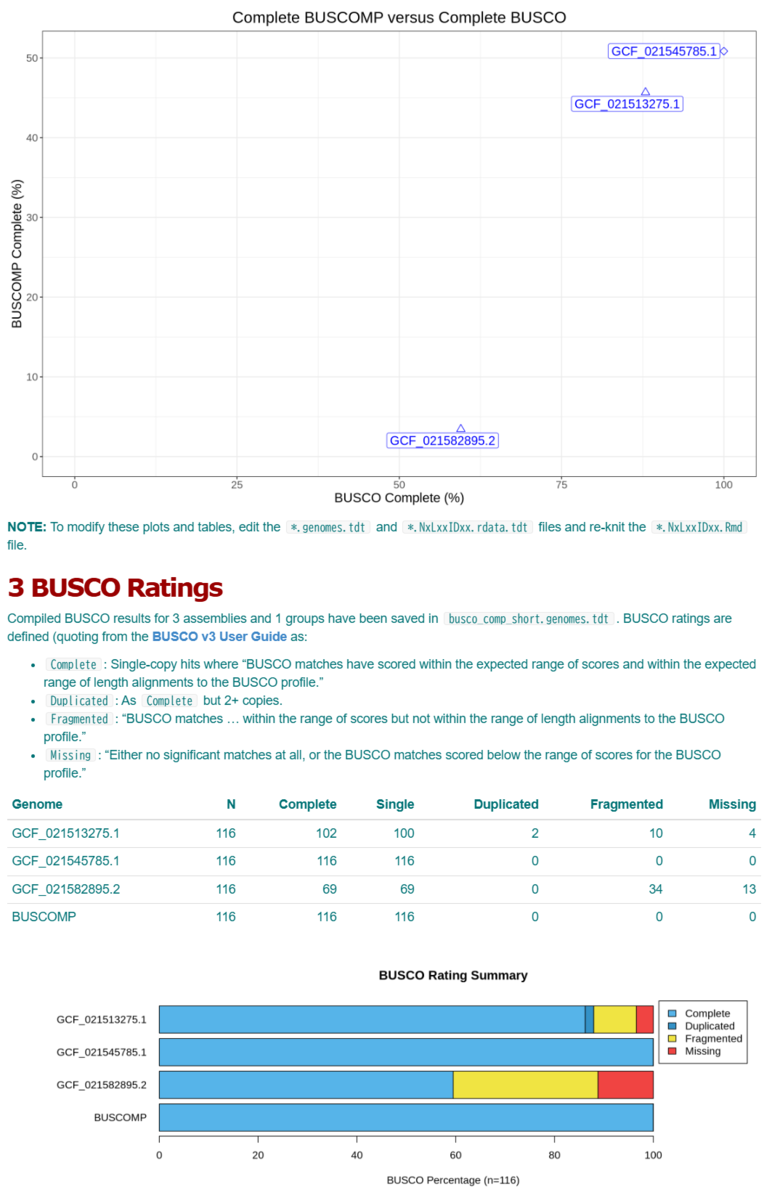




補足
- <スライドより>BUSCOの結果は、アセンブリの構造や品質に敏感であり、同じゲノムの異なるアセンブリ間で比較する際に一貫性を欠くことがある。BUSCOMPは、これらの課題を解決するために開発されている。
- 具体的には、複数のアセンブリから最高スコアの完全なBUSCO遺伝子配列を集めて、これらを基準として各アセンブリにマッピングする。これを行うのは、BUSCOは各アセンブリに対して個別に実行されるため、同じ遺伝子でも、あるアセンブリでは完全、一方では欠損として報告されることがあるため、 どのBUSCOが共通で、どれが検出されなかったのかを比較できないからである。
- 全体で見つかったBUSCO遺伝子の統一リストを作り、そのリストに含まれる各BUSCO遺伝子を、各アセンブリの配列にBLASTなどでマッピングし直す。そして各アセンブリがその共通セットのうちいくつを保持しているかを再評価する。このプロセスにより、アセンブリ間で正確に比較をできるようにしている。
引用
BUSCOMP: BUSCO compilation and comparison – Assessing completeness in multiple genome assemblies
Richard J. Edwards
Published 01 Jul 2019 (https://doi.org/10.7490/f1000research.1116972.1)
BUSCOMPレポジトリにはこちら ↓ を引用するように記載されている。
Transcript- and annotation-guided genome assembly of the European starling
Katarina C Stuart 1, Richard J Edwards 2, Yuanyuan Cheng 3, Wesley C Warren 4, David W Burt 5, William B Sherwin 1, Natalie R Hofmeister 6 7, Scott J Werner 8, Gregory F Ball 9, Melissa Bateson 10, Matthew C Brandley 11, Katherine L Buchanan 12, Phillip Cassey 13, David F Clayton 14, Tim De Meyer 15, Simone L Meddle 16, Lee A Rollins 1 12
Mol Ecol Resour. 2022 Nov;22(8):3141-3160. doi: 10.1111/1755-0998.13679. Epub 2022 Jul 18.
関連