2024/06/24 誤字修正
2020年の論文
IQ-TREE(http://www.iqtree.org)は、最尤法を用いた系統推論を行うための、ユーザーフレンドリーで広く利用されているソフトウェアパッケージである。2014年にバージョン1をリリースして以来、本著者らはIQ-TREEを継続的に拡張し、ゲノムデータを扱うための配列進化の新しいモデルや系統推定の効率的な計算アプローチを多数統合してきた。ここでは、IQ-TREEバージョン2の特筆すべき機能について説明し、他のソフトウェアと比較した際の主な利点を強調する。
2015年の論文
大規模な系統学データセットでは、特に最尤(ML)系統樹のための高速な樹推定法が必要とされる。高速なプログラムも存在するが、最適な樹形を見つけるinherent heuristicsのため、最適なツリーが見つかっているかどうかは明らかではない。従って、MLツリーを見つけるために異なる探索戦略を採用し、同時に現在利用可能なMLプログラムと同程度に高速なアプローチを追加する必要がある。本著者らは、ヒルクライムアプローチとstochastic perturbation methodの組み合わせが時間効率よく実装できることを示す。RAxMLやPhyMLと同じCPU時間であれば、IQ-TREEは62.2%から87.1%より高い尤度を発見し、効率的にツリー空間を探索した。IQ-TREEの停止ルールを用いた場合、DNAアラインメントでは75.7%、47.1%、タンパク質アラインメントでは42.2%、100%でRAxMLとPhyMLの方が高速となる。しかし、IQ-TREEにより高い尤度が得られる範囲は73.3-97.1%に改善された。
Manual
http://www.iqtree.org/doc/
Beginner's Tutorial
http://www.iqtree.org/doc/Tutorial
Advanced Tutorial
http://www.iqtree.org/doc/Advanced-Tutorial
コマンドリファレンス
http://www.iqtree.org/doc/Command-Reference
v1系とv2以降ではコマンドが微妙に異なっています。ここではv2を簡単に紹介します。
インストール
Github
https://github.com/iqtree/iqtree2
#link(iqtreeと指定すると通常v2が導入される)
mamba install bioconda::iqtree -y
HPからもそれぞれのプラットフォームのバージョンのIQ-TREEをダウンロードできる。2024年現在v2.3.4となっている。

アクセスして、一番下にスクロールすると全プラットフォームのダウンロードリンクがある。
> iqtree2 -h
IQ-TREE multicore version 2.3.4 COVID-edition for Linux x86 64-bit built Jun 5 2024
Developed by Bui Quang Minh, James Barbetti, Nguyen Lam Tung, Olga Chernomor,
Heiko Schmidt, Dominik Schrempf, Michael Woodhams, Ly Trong Nhan, Thomas Wong
Usage: iqtree [-s ALIGNMENT] [-p PARTITION] [-m MODEL] [-t TREE] ...
GENERAL OPTIONS:
-h, --help Print (more) help usages
-s FILE[,...,FILE] PHYLIP/FASTA/NEXUS/CLUSTAL/MSF alignment file(s)
-s DIR Directory of alignment files
--seqtype STRING BIN, DNA, AA, NT2AA, CODON, MORPH (default: auto-detect)
-t FILE|PARS|RAND Starting tree (default: 99 parsimony and BIONJ)
-o TAX[,...,TAX] Outgroup taxon (list) for writing .treefile
--prefix STRING Prefix for all output files (default: aln/partition)
--seed NUM Random seed number, normally used for debugging purpose
--safe Safe likelihood kernel to avoid numerical underflow
--mem NUM[G|M|%] Maximal RAM usage in GB | MB | %
--runs NUM Number of indepedent runs (default: 1)
-v, --verbose Verbose mode, printing more messages to screen
-V, --version Display version number
--quiet Quiet mode, suppress printing to screen (stdout)
-fconst f1,...,fN Add constant patterns into alignment (N=no. states)
--epsilon NUM Likelihood epsilon for parameter estimate (default 0.01)
-T NUM|AUTO No. cores/threads or AUTO-detect (default: 1)
--threads-max NUM Max number of threads for -T AUTO (default: all cores)
CHECKPOINT:
--redo Redo both ModelFinder and tree search
--redo-tree Restore ModelFinder and only redo tree search
--undo Revoke finished run, used when changing some options
--cptime NUM Minimum checkpoint interval (default: 60 sec and adapt)
PARTITION MODEL:
-p FILE|DIR NEXUS/RAxML partition file or directory with alignments
Edge-linked proportional partition model
-q FILE|DIR Like -p but edge-linked equal partition model
-Q FILE|DIR Like -p but edge-unlinked partition model
-S FILE|DIR Like -p but separate tree inference
--subsample NUM Randomly sub-sample partitions (negative for complement)
--subsample-seed NUM Random number seed for --subsample
LIKELIHOOD/QUARTET MAPPING:
--lmap NUM Number of quartets for likelihood mapping analysis
--lmclust FILE NEXUS file containing clusters for likelihood mapping
--quartetlh Print quartet log-likelihoods to .quartetlh file
TREE SEARCH ALGORITHM:
--ninit NUM Number of initial parsimony trees (default: 100)
--ntop NUM Number of top initial trees (default: 20)
--nbest NUM Number of best trees retained during search (defaut: 5)
-n NUM Fix number of iterations to stop (default: OFF)
--nstop NUM Number of unsuccessful iterations to stop (default: 100)
--perturb NUM Perturbation strength for randomized NNI (default: 0.5)
--radius NUM Radius for parsimony SPR search (default: 6)
--allnni Perform more thorough NNI search (default: OFF)
-g FILE (Multifurcating) topological constraint tree file
--fast Fast search to resemble FastTree
--polytomy Collapse near-zero branches into polytomy
--tree-fix Fix -t tree (no tree search performed)
--treels Write locally optimal trees into .treels file
--show-lh Compute tree likelihood without optimisation
--terrace Check if the tree lies on a phylogenetic terrace
ULTRAFAST BOOTSTRAP/JACKKNIFE:
-B, --ufboot NUM Replicates for ultrafast bootstrap (>=1000)
-J, --ufjack NUM Replicates for ultrafast jackknife (>=1000)
--jack-prop NUM Subsampling proportion for jackknife (default: 0.5)
--sampling STRING GENE|GENESITE resampling for partitions (default: SITE)
--boot-trees Write bootstrap trees to .ufboot file (default: none)
--wbtl Like --boot-trees but also writing branch lengths
--nmax NUM Maximum number of iterations (default: 1000)
--nstep NUM Iterations for UFBoot stopping rule (default: 100)
--bcor NUM Minimum correlation coefficient (default: 0.99)
--beps NUM RELL epsilon to break tie (default: 0.5)
--bnni Optimize UFBoot trees by NNI on bootstrap alignment
NON-PARAMETRIC BOOTSTRAP/JACKKNIFE:
-b, --boot NUM Replicates for bootstrap + ML tree + consensus tree
-j, --jack NUM Replicates for jackknife + ML tree + consensus tree
--jack-prop NUM Subsampling proportion for jackknife (default: 0.5)
--bcon NUM Replicates for bootstrap + consensus tree
--bonly NUM Replicates for bootstrap only
--tbe Transfer bootstrap expectation
SINGLE BRANCH TEST:
--alrt NUM Replicates for SH approximate likelihood ratio test
--alrt 0 Parametric aLRT test (Anisimova and Gascuel 2006)
--abayes approximate Bayes test (Anisimova et al. 2011)
--lbp NUM Replicates for fast local bootstrap probabilities
MODEL-FINDER:
-m TESTONLY Standard model selection (like jModelTest, ProtTest)
-m TEST Standard model selection followed by tree inference
-m MF Extended model selection with FreeRate heterogeneity
-m MFP Extended model selection followed by tree inference
-m ...+LM Additionally test Lie Markov models
-m ...+LMRY Additionally test Lie Markov models with RY symmetry
-m ...+LMWS Additionally test Lie Markov models with WS symmetry
-m ...+LMMK Additionally test Lie Markov models with MK symmetry
-m ...+LMSS Additionally test strand-symmetric models
--mset STRING Restrict search to models supported by other programs
(raxml, phyml, mrbayes, beast1 or beast2)
--mset STR,... Comma-separated model list (e.g. -mset WAG,LG,JTT)
--msub STRING Amino-acid model source
(nuclear, mitochondrial, chloroplast or viral)
--mfreq STR,... List of state frequencies
--mrate STR,... List of rate heterogeneity among sites
(e.g. -mrate E,I,G,I+G,R is used for -m MF)
--cmin NUM Min categories for FreeRate model [+R] (default: 2)
--cmax NUM Max categories for FreeRate model [+R] (default: 10)
--merit AIC|AICc|BIC Akaike|Bayesian information criterion (default: BIC)
--mtree Perform full tree search for every model
--madd STR,... List of mixture models to consider
--mdef FILE Model definition NEXUS file (see Manual)
--modelomatic Find best codon/protein/DNA models (Whelan et al. 2015)
PARTITION-FINDER:
--merge Merge partitions to increase model fit
--merge greedy|rcluster|rclusterf
Set merging algorithm (default: rclusterf)
--merge-model 1|all Use only 1 or all models for merging (default: 1)
--merge-model STR,...
Comma-separated model list for merging
--merge-rate 1|all Use only 1 or all rate heterogeneity (default: 1)
--merge-rate STR,...
Comma-separated rate list for merging
--rcluster NUM Percentage of partition pairs for rcluster algorithm
--rclusterf NUM Percentage of partition pairs for rclusterf algorithm
--rcluster-max NUM Max number of partition pairs (default: 10*partitions)
SUBSTITUTION MODEL:
-m STRING Model name string (e.g. GTR+F+I+G)
DNA: HKY (default), JC, F81, K2P, K3P, K81uf, TN/TrN, TNef,
TIM, TIMef, TVM, TVMef, SYM, GTR, or 6-digit model
specification (e.g., 010010 = HKY)
Protein: LG (default), Poisson, cpREV, mtREV, Dayhoff, mtMAM,
JTT, WAG, mtART, mtZOA, VT, rtREV, DCMut, PMB, HIVb,
HIVw, JTTDCMut, FLU, Blosum62, GTR20, mtMet, mtVer, mtInv, FLAVI,
Q.LG, Q.pfam, Q.pfam_gb, Q.bird, Q.mammal, Q.insect, Q.plant, Q.yeast
Protein mixture: C10,...,C60, EX2, EX3, EHO, UL2, UL3, EX_EHO, LG4M, LG4X
Binary: JC2 (default), GTR2
Empirical codon: KOSI07, SCHN05
Mechanistic codon: GY (default), MG, MGK, GY0K, GY1KTS, GY1KTV, GY2K,
MG1KTS, MG1KTV, MG2K
Semi-empirical codon: XX_YY where XX is empirical and YY is mechanistic model
Morphology/SNP: MK (default), ORDERED, GTR
Lie Markov DNA: 1.1, 2.2b, 3.3a, 3.3b, 3.3c, 3.4, 4.4a, 4.4b, 4.5a,
4.5b, 5.6a, 5.6b, 5.7a, 5.7b, 5.7c, 5.11a, 5.11b, 5.11c,
5.16, 6.6, 6.7a, 6.7b, 6.8a, 6.8b, 6.17a, 6.17b, 8.8,
8.10a, 8.10b, 8.16, 8.17, 8.18, 9.20a, 9.20b, 10.12,
10.34, 12.12 (optionally prefixed by RY, WS or MK)
Non-reversible: STRSYM (strand symmetric model, equiv. WS6.6),
NONREV, UNREST (unrestricted model, equiv. 12.12)
NQ.pfam, NQ.bird, NQ.mammal, NQ.insect, NQ.plant, NQ.yeast
Otherwise: Name of file containing user-model parameters
STATE FREQUENCY:
-m ...+F Empirically counted frequencies from alignment
-m ...+FO Optimized frequencies by maximum-likelihood
-m ...+FQ Equal frequencies
-m ...+FRY For DNA, freq(A+G)=1/2=freq(C+T)
-m ...+FWS For DNA, freq(A+T)=1/2=freq(C+G)
-m ...+FMK For DNA, freq(A+C)=1/2=freq(G+T)
-m ...+Fabcd 4-digit constraint on ACGT frequency
(e.g. +F1221 means f_A=f_T, f_C=f_G)
-m ...+FU Amino-acid frequencies given protein matrix
-m ...+F1x4 Equal NT frequencies over three codon positions
-m ...+F3x4 Unequal NT frequencies over three codon positions
RATE HETEROGENEITY AMONG SITES:
-m ...+I A proportion of invariable sites
-m ...+G[n] Discrete Gamma model with n categories (default n=4)
-m ...*G[n] Discrete Gamma model with unlinked model parameters
-m ...+I+G[n] Invariable sites plus Gamma model with n categories
-m ...+R[n] FreeRate model with n categories (default n=4)
-m ...*R[n] FreeRate model with unlinked model parameters
-m ...+I+R[n] Invariable sites plus FreeRate model with n categories
-m ...+Hn Heterotachy model with n classes
-m ...*Hn Heterotachy model with n classes and unlinked parameters
--alpha-min NUM Min Gamma shape parameter for site rates (default: 0.02)
--gamma-median Median approximation for +G site rates (default: mean)
--rate Write empirical Bayesian site rates to .rate file
--mlrate Write maximum likelihood site rates to .mlrate file
POLYMORPHISM AWARE MODELS (PoMo):
-s FILE Input counts file (see manual)
-m ...+P DNA substitution model (see above) used with PoMo
-m ...+N<POPSIZE> Virtual population size (default: 9)
-m ...+WB|WH|S] Weighted binomial sampling
-m ...+WH Weighted hypergeometric sampling
-m ...+S Sampled sampling
-m ...+G[n] Discrete Gamma rate with n categories (default n=4)
COMPLEX MODELS:
-m "MIX{m1,...,mK}" Mixture model with K components
-m "FMIX{f1,...fK}" Frequency mixture model with K components
--mix-opt Optimize mixture weights (default: detect)
-m ...+ASC Ascertainment bias correction
--tree-freq FILE Input tree to infer site frequency model
--site-freq FILE Input site frequency model file
--freq-max Posterior maximum instead of mean approximation
TREE TOPOLOGY TEST:
--trees FILE Set of trees to evaluate log-likelihoods
--test NUM Replicates for topology test
--test-weight Perform weighted KH and SH tests
--test-au Approximately unbiased (AU) test (Shimodaira 2002)
--sitelh Write site log-likelihoods to .sitelh file
ANCESTRAL STATE RECONSTRUCTION:
--ancestral Ancestral state reconstruction by empirical Bayes
--asr-min NUM Min probability of ancestral state (default: equil freq)
TEST OF SYMMETRY:
--symtest Perform three tests of symmetry
--symtest-only Do --symtest then exist
--symtest-remove-bad Do --symtest and remove bad partitions
--symtest-remove-good Do --symtest and remove good partitions
--symtest-type MAR|INT Use MARginal/INTernal test when removing partitions
--symtest-pval NUMER P-value cutoff (default: 0.05)
--symtest-keep-zero Keep NAs in the tests
CONCORDANCE FACTOR ANALYSIS:
-t FILE Reference tree to assign concordance factor
--gcf FILE Set of source trees for gene concordance factor (gCF)
--df-tree Write discordant trees associated with gDF1
--scf NUM Number of quartets for site concordance factor (sCF)
--scfl NUM Like --scf but using likelihood (recommended)
-s FILE Sequence alignment for --scf
-p FILE|DIR Partition file or directory for --scf
--cf-verbose Write CF per tree/locus to cf.stat_tree/_loci
--cf-quartet Write sCF for all resampled quartets to .cf.quartet
ALISIM: ALIGNMENT SIMULATOR
Usage: iqtree --alisim <OUTPUT_PREFIX> [-m MODEL] [-t TREE] ...
--alisim OUTPUT_ALIGNMENT Activate AliSim and specify the output alignment filename
-t TREE_FILE Set the input tree file name
--length LENGTH Set the length of the root sequence
--num-alignments NUMBER Set the number of output datasets
--seqtype STRING BIN, DNA, AA, CODON, MORPH{NUM_STATES} (default: auto-detect)
For morphological data, 0<NUM_STATES<=32
--m MODEL_STRING Specify the evolutionary model. See Manual for more detail
--mdef FILE Name of a NEXUS model file to define new models (see Manual)
--fundi TAXA_LIST,RHO Specify a list of taxa, and Rho (Fundi weight) for FunDi model
--indel <INS>,<DEL> Set the insertion and deletion rate of the indel model,
relative to the substitution rate
--indel-size <INS_DIS>,<DEL_DIS> Set the insertion and deletion size distributions
--sub-level-mixture Enable the feature to simulate substitution-level mixture model
--no-unaligned Disable outputing a file of unaligned sequences
when using indel models
--root-seq FILE,SEQ_NAME Specify the root sequence from an alignment
-s FILE Specify the input sequence alignment
--no-copy-gaps Disable copying gaps from input alignment (default: false)
--site-freq <OPTION> Specify the option (MEAN (default), or SAMPLING, or MODEL)
to mimic the site-frequencies for mixture models from
the input alignment (see Manual)
--site-rate <OPTION> Specify the option (MEAN (default), or SAMPLING, or MODEL)
to mimic the discrete rate heterogeneity from
the input alignment (see Manual)
-t RANDOM{MODEL,NUM_TAXA} Specify the model and the number of taxa to generate a random tree
-rlen MIN MEAN MAX Specify three numbers: minimum, mean and maximum branch lengths
when generating a random tree
-p FILE NEXUS/RAxML partition file
Edge-linked proportional partition model
-q FILE Like -p but edge-linked equal partition model
-Q FILE Like -p but edge-unlinked partition model
--distribution FILE Supply a definition file of distributions,
which could be used to generate random model parameters
--branch-distribution DIS Specify a distribution, from which branch lengths of the input trees
are randomly generated and overridden.
--branch-scale SCALE Specify a value to scale all branch lengths
--single-output Output all alignments into a single file
--write-all Enable outputting internal sequences
--seed NUM Random seed number (default: CPU clock)
Be careful to make the AliSim reproducible,
users should specify the seed number
-gz Enable output compression but taking longer running time
-af phy|fasta Set the output format (default: phylip)
User Manual is available at http://www.iqtree.org/doc/alisim
ANALYSIS WITH GENTRIUS ALGORITHM:
--gentrius FILE File must contain either a single species-tree or a set of subtrees.
-pr_ab_matrix FILE Presence-absence matrix of loci coverage.
-s FILE PHYLIP/FASTA/NEXUS/CLUSTAL/MSF alignment file(s)
-p FILE NEXUS/RAxML partition file
-g_stop_t NUM Stop after NUM species-trees were generated, or use 0 to turn off this stopping rule. Default: 1MLN trees.
-g_stop_i NUM Stop after NUM intermediate trees were visited, or use 0 to turn off this stopping rule. Default: 10MLN trees.
-g_stop_h NUM Stop after NUM hours (CPU time), or use 0 to turn off this stopping rule. Default: 7 days.
-g_non_stop Turn off all stopping rules.
-g_query FILE Species-trees to test for identical set of subtrees.
-g_print Write all generated species-trees. WARNING: there might be millions of trees!
-g_print_lim NUM Limit on the number of species-trees to be written.
-g_print_induced Write induced partition subtrees.
-g_print_m Write presence-absence matrix.
-g_rm_leaves NUM Invoke reverse analysis for complex datasets.
TIME TREE RECONSTRUCTION:
--date FILE File containing dates of tips or ancestral nodes
--date TAXNAME Extract dates from taxon names after last '|'
--date-tip STRING Tip dates as a real number or YYYY-MM-DD
--date-root STRING Root date as a real number or YYYY-MM-DD
--date-ci NUM Number of replicates to compute confidence interval
--clock-sd NUM Std-dev for lognormal relaxed clock (default: 0.2)
--date-no-outgroup Exclude outgroup from time tree
--date-outlier NUM Z-score cutoff to remove outlier tips/nodes (e.g. 3)
--date-options ".." Extra options passing directly to LSD2
--dating STRING Dating method: LSD for least square dating (default)
実行方法
実行するには、多重整列されたPHYLIPフォーマットかFASTAフォーマット、あるいはNEXUS/CLUSTAL/MSFの配列を準備する必要がある。

IQ-treeチュートリアルより
IQ-TREEを実行するには、多重整列された配列セットを指定する。オプションなしだと、デフォルト設定での最尤推定が行われる。
#PHYLIP
iqtree2 -s example.phy
#FASTA
iqtree2 -s example.fa
- -s PHYLIP/FASTA/NEXUS/CLUSTAL/MSF alignment file(s)
- --seqtype BIN, DNA, AA, NT2AA, CODON, MORPH (default: auto-detect)
置換モデルを選択する。
iqtree2 -s example.phy -m GTR+G
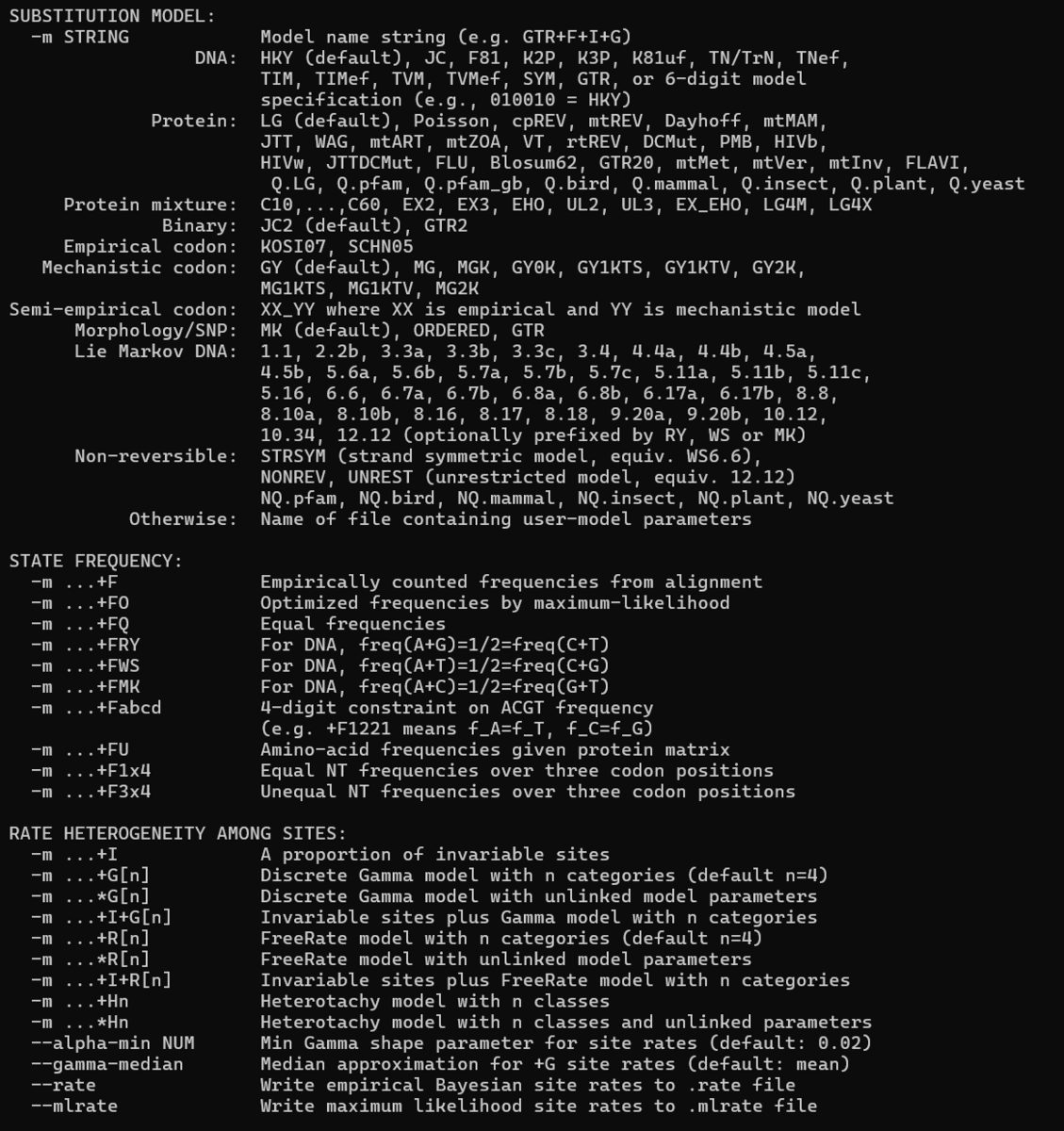
Substitution models
http://www.iqtree.org/doc/Substitution-Models
データに対してどのモデルが適切かわからない場合は、ModelFinder を使用して最適なモデルを選択できる。ModelFinder Plus : MFPは、AIC(Akaike Information Criterion)、BIC(Bayesian Information Criterion)に基づいて、モデルの適合度と複雑さを評価し、最適な置換モデルを選択する。
iqtree2 -s example.phy -m MFP

ModelFinder Plusを使ってデータに最適なモデルを選び、近似的に最適なモデルを組み合わせてさらにデータに適合する進化モデルを作成する(近いモデルを統合して新しい複合モデルを作成)
iqtree2 -s example.phy -m MFP+MERGE
すべてのモデルについて完全なツリー推論を行う(高負荷)
iqtree -s example.phy -m MFP+MERGE --mtree
- --mtree Perform full tree search for every model
最適なスレッド数を自動で決定、ただし割り当てられるCPUコアの上限は32に設定。
iqtree -s example.phy -T AUTO --threads-max 32
- -T No. cores/threads or AUTO-detect (default: 1)
- --threads-max Max number of threads for -T AUTO (default: all cores)
ブートストラップ法を実行。
iqtree2 –s example.phy -b 100
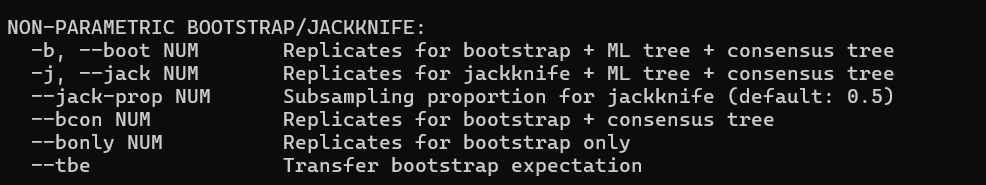
ランダムにポジション列を選択後、ノンパラメトリックに初期ツリー構築から計算が繰り返されるのでかなり時間がかかる。
超高速ブートストラップ近似を実行(UFBoot 2は標準的な手順よりも桁違いに速い)。
iqtree2 –s example.phy –B 1000 #v1系なら-bb 1000

UFBootでは、支持率が≧ 95%の場合のみ、そのクレードを信じ始めるべき。BS%とUFBoot%を直接比較すべきではない(マニュアルより)。
(モデルが収束するまで繰り返し、1000を指定したなら最大1000回ということ)
-alrt(Approximate Likelihood-Ratio Test)を追加して枝の支持率を評価する。aLRTを1000回繰り返す。
iqtree -s example.phy -B 1000 -alrt 1000
SH like aLRT検定(paper)では、各ブランチには、SH-aLRTとUFBootのサポートが割り当てられる。通常、SH-aLRT >= 80%、UFboot >= 95%であれば、そのクレードを信頼し始めてよい(マニュアルより)。
深刻なモデル違反によるブランチサポートの過大評価を防ぐため、各ブートストラップツリーをさらに最適化
iqtree -s example.phy -B 1000 -bnni
アウトグループを指定
iqtree -s example.phy –o Lizard
(IQ-TREEは与えられた配列の生物学的背景について何も知らない。IQ-TREEは、アラインメントで最初に出現する配列をroot部分にツリーを描画する)
全プロセスをやり直す。
iqtree -s example.phy --redo

全プロセスを独立に複数回実行する。-vでよりたくさんのログメッセージを表示する。
iqtree -s example.phy --runs 3 -v
- --runs Number of indepedent runs (default: 1)
- -v Verbose mode, printing more messages to screen
IQ-TREEは自動で樹形推定プロセスを進めるため、ログを読むことが重要(.logとしてファイル保存されている)
勉強会用の資料を貼っておきます。



IQ-treeにはDiscussion forumが作成されており(old version)、そこでたくさんディスカッションされています。
https://github.com/iqtree/iqtree2/discussions
コメント
IQ-treeは樹形推定で煩雑なプロセスを自動で高速実行でき、人気のツールとなっていますが、それゆえ知識がないと現在の生命史とは矛盾するようなツリーも作りがちです。ログを読んだり樹形の信頼性を見るのはもちろんのこと、先行研究とよく比較する事も重要かと思います。遺伝子のツリーでも生命のツリーでも同様です。
その他
- 定期的にチェックポイントファイル example.phy.ckp.gzがディスクに書き込まれる。このチェックポイントファイルは、中断された実行を再開するために使用される。
- IQtreeは実行されると最初にアライメント内のすべての配列について組成カイ二乗検定を実行する。その目的は、文字構成(DNAの場合はヌクレオチド、タンパク質配列の場合はアミノ酸など)の均一性をテストすることにある(#197)。文字組成がアラインメントの平均組成から著しく逸脱している場合、その配列は不合格と判定される(例;sequences failed composition chi2 test (p-value<5%; df=3))。この検定は、データセットの問題点を突き止めるのに役立つ探索的なツールとして組み込まれている。通常、デフォルトでは失敗した配列を削除することはないだろうが、もしツリーが予期しないトポロジーを示すなら、この検定は問題の起源の方向を示すかもしれない。パーティションによる複数遺伝子からなるデータがある場合、パーティション分析で各遺伝子のパーティションごとに別々にテストする方が合理的かもしれない(遺伝子によって異なる進化の歴史や特徴を持っている可能性画あるという意味と考えられる)。タンパク質データである場合、配列に沿って異なるアミノ酸組成を考慮したいくつかのタンパク質混合モデル、例えばC10からC60プロファイル混合モデルを試すこともできる(こちらからそのまま引用)。
引用
IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era
Bui Quang Minh, Heiko A Schmidt, Olga Chernomor, Dominik Schrempf, Michael D Woodhams, Arndt von Haeseler, Robert Lanfear
Molecular Biology and Evolution, Volume 37, Issue 5, May 2020, Pages 1530–1534
IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies
Lam-Tung Nguyen, Heiko A. Schmidt, Arndt von Haeseler, and Bui Quang Minh
Molecular Biology and Evolution. 2015 Jan; 32(1): 268–274.
ModelFinder: fast model selection for accurate phylogenetic estimates
Subha Kalyaanamoorthy, Bui Quang Minh, Thomas K F Wong, Arndt von Haeseler & Lars S Jermiin
Nature Methods volume 14, pages587–589 (2017)
Ultrafast Approximation for Phylogenetic Bootstrap
Bui Quang Minh, Minh Anh Thi Nguyen, Arndt von Haeseler
Molecular Biology and Evolution, Volume 30, Issue 5, May 2013, Pages 1188–1195
参考
http://www.tezuru-mozuru.com/?cat=200