ユニバーサルシングルコピーオルソログは、ゲノムの最も保存された構成要素である。これらは進化の歴史の研究や新しいアセンブリの評価に日常的に使用されているが、現在の方法は利用可能なゲノムデータからの情報を組み込んでいない。本研究ではまず、進化史が普遍的遺伝子内容に与える影響を分析し、植物、真菌、動物の11,098ゲノム(2,606の分類群)において、215の分類群がそれぞれの系統群と比較してBUSCO(Benchmarking Universal Single Copy Orthologs)の完全性において有意な差異を示すことを明らかにした。さらに、169のグループでは、祖先の全ゲノム重複イベントから由来する可能性のある重複オルソログ数が著しく増加していた。次に、広範なBUSCO由来の系統樹における分類学的整合性の程度を調査した。543のテスト対象の科のうち275のproperな科において、高い進化速度を示すサイトは、低い進化速度のサイトと比較して、最大23.84%多くの分類学的に一致した系統樹を生成し、少なくとも46.15%少ない末端変異性の高い系統樹を生成することが判明した。本著者らは、BUSCO連結ツリーと共役ツリーが同等の精度を有することを発見し、連結アラインメントから得られる高い進化速度のサイトが、最も一致性が高く、最も変動の少ない系統樹を生成すると結論付けた。最後に、検出されないが広範なBUSCO遺伝子喪失イベントが、アセンブリ品質の誤った表現を引き起こすことを示した。これに対処するため、標準の方法と比較して最大6.99%少ない偽陽性を提供するキュレーションされたBUSCOセット(CUSCOs)をフィルタリングし、遺伝子シントニーを用いたアセンブリ比較のための新規手法を導入した。アセンブリ評価において進化の歴史を考慮することの重要性を強調し、一貫した系統樹を再構築し、より正確なアセンブリ評価を提供するphycaソフトウェアツールキットをリリースする。
phyca

(上のボタンから分類群の切り替えが可能)
インストール
compleasmが依存しているdendropy(系統樹解析のライブラリ)の古いバージョンを要求するが、dendropyは新しいpython環境に対応していない。レポジトリの指定通り、compleasmをpython3.9の環境にインストールし、それからphycaをpipで導入した。
mamba create -n phyca python=3.9.26 -y
conda activate phyca
mamba install bioconda::compleasm=0.2.6 -y
#本体
pip install phyca
$ phyca
/home/kazu/mambaforge/envs/compleasm_env/lib/python3.9/site-packages/phyca/phyca.py:9: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import resource_filename
usage: phyca [-h] [-v] [-a assembly] [-c compleasm_directory] -l lineage [-o output] [-t threads] [-r reference] [-m rcompleasm_directory] [-n] [-s] [-i] [-d] [-w]
phyca
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version number and exit
-a assembly, --assembly assembly
Assembly in FASTA format
-c compleasm_directory, --compdir compleasm_directory
Compleasm output directory
BUSCO lineage
-o output, --output output
Output prefix
-t threads, --threads threads
Compleasm threads
-r reference, --reference reference
Reference assembly
-m rcompleasm_directory, --rcompdir rcompleasm_directory
Reference compleasm output directory
-n, --nullify Remove all BUSCO genes in assembly
-s, --syndis Compute syntenic distance from reference
-i, --ignore_orientation
Ignores orientation and only considers gene order for syntenic distances.
-d, --include_duplications
Duplicated gene pairs are considered distinct for syntenic distances.
-w, --include_singleton_contigs
Includes contigs with single genes for syntenic distances.
実行方法
phyca は、Compleasm をベースに構築されたツールで、BUSCO や Compleasm によるアノテーション精度を最大7%向上させ、公開されている参照ゲノムとのシンテニー比較、さらに事前計算された系統樹への配置を迅速に行う。
実行するには、クエリとなるゲノムアセンブリを指定する。以下の系統がサポートされている。['arthropoda', 'ascomycota', 'basidiomycota', 'chlorophyta', 'eudicots', 'fungi', 'liliopsida', 'metazoa', 'vertebrata', 'viridiplantae']
phyca -a input_assembly.fna -l metazoa -t 20
既にCompleasm の結果を保持している場合、Compleasm の出力ディレクトリを指定することもできる。
phyca -c compleasm_dir/ -l <lineage>
Rattus norvegicusゲノムを使用したところ、計算が終わるまで30分ほどかかった(5995WX, 20スレッド)。
出力例

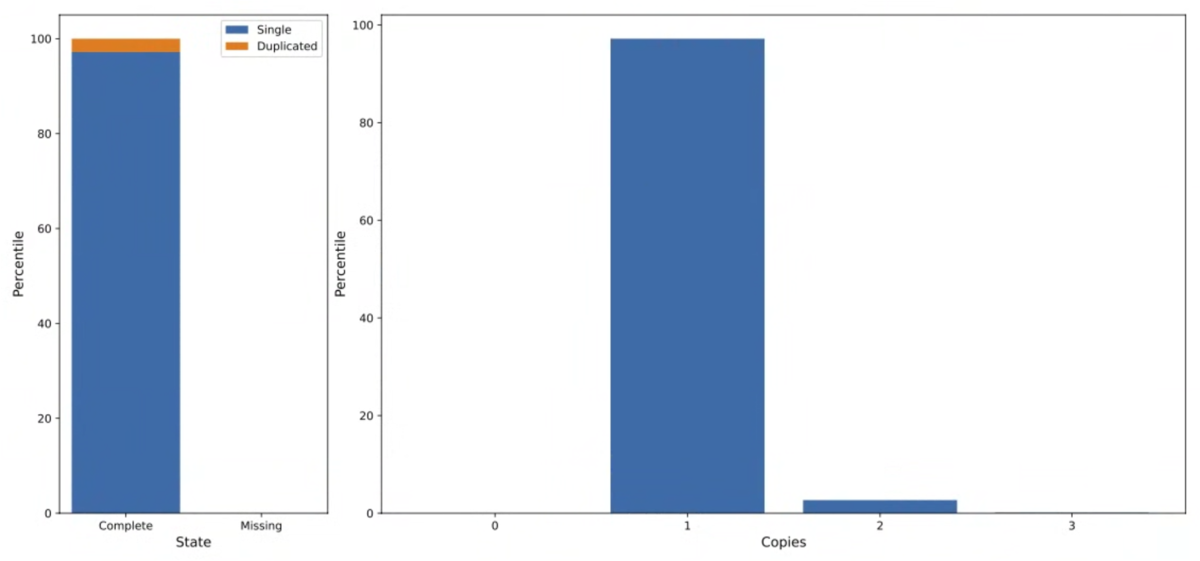
BUSCO、CUSCO(精度が向上したキュレーション済みのUniversal Single Copy Orthologs)およびMUSCO(残りのUniversal Single Copy Orthologs)の統計データとグラフが出力される。系統樹も作成されるが、テスト時は最終ステップである系統樹作成時、Bionick関連のエラーが出て正常に終わらないデータもあった。
USCO_stats.tsv

USCO_copies.tsv

USCO_bars.pdf
SynIdentity.tsv
synteny_tree_dashed.pdf

複数のゲノムアセンブリ間のCompleasm出力を比較する機能も用意されている。2つのゲノムを指定するか、2つのゲノムそれぞれのを指定する。
#assemnblyを指定
phyca -l <lineage> -s -a assembly1.fna -r assembly2.fna
#Compleasmを指定
phyca -l <lineage> -s -c compleasm_dir1 -m compleasm_dir2
(論文より)
- 10 の主要 BUSCO 真核生物系統(緑色植物、単子葉類、双子葉類、緑藻、真菌、子嚢菌、担子菌、後生動物、節足動物、脊椎動物)に対してより高い特異性を達成する、キュレートされた BUSCO オルソログセット(CUSCOs)を整備した。近縁アセンブリの堅牢な比較と評価のために、標準的な BUSCO 検索よりも高いコントラストと解像度を提供するシンテニー BUSCO 指標を導出した。
引用
Universal orthologs infer deep phylogenies and improve genome quality assessments
Md Nafis Ul Alam, Cristian Román-Palacios, Dario Copetti & Rod A. Wing
BMC Biology volume 23, Article number: 224 (2025)
関連