メタゲノムデータにおける微生物の分類群と相対的豊度の決定は、技術的に依然として困難である。本研究では、普遍的マーカー遺伝子内の保存領域を用いて群集構成を推定する「SingleM」を提案する。ゲノム情報が欠如した種を正確に組み込むことで、未知種がほとんどの環境微生物群集で優勢であることを示した。当研究チームが運営するウェブサイト「Sandpiper」は、公開されている248,559件のメタゲノムデータから微生物群集プロファイルを収集している。
SingleMについては以前紹介しています(古い可能性があるのでレポジトリを確認してください)。
ここではSandpiperのwebサイトを簡単に見ていきます。
About
https://sandpiper.qut.edu.au/About
https://sandpiper.qut.edu.au/にアクセスする。

分類学的プロファイリングツールとしての SingleM の有用性を利用して、NCBI SRAに登録されているメタゲノムデータのうち、2021年12月時点までで公開されていたものに適用し、合計 248,559 件のメタゲノム(17,617 プロジェクトにわたる)からコミュニティプロファイルを生成した。これらは 総シーケンス量 1.3 Pbp(ペタ塩基対) に相当する。Sandpiper ウェブサイトではこの大規模解析の結果を公開している。
メニューのSearchから検索できる。検索は、GTDB taxonomyやサンプルのシークエンスアーカイブのアクセッションなどを使って行う。

出力を見てみる。
Example1: sponge microbiome (PRJNA555144 | SRR9841429)
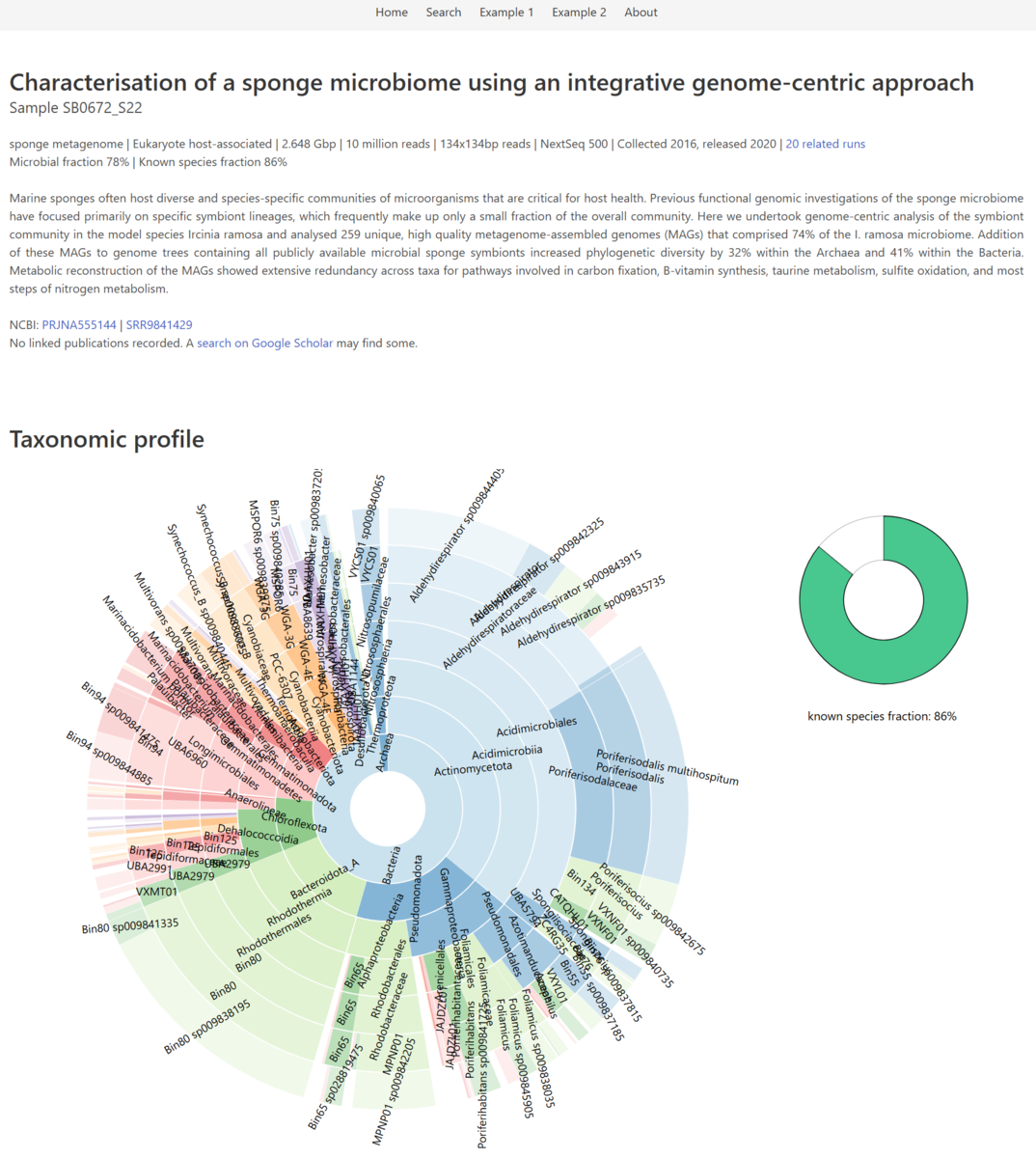
SingleMによって各マーカー遺伝子から生成された OTU テーブルは、1つの分類学的プロファイルに統合され、各分類群ごとのリードカバレッジがメタゲノム内でどの程度あるかを表す単一のプロファイルが生成される。各分類群の相対存在量は、各分類群のリードカバレッジをメタゲノム全体のリードカバレッジで割ることによって算出される。Sandpiper の可視化にはこの相対存在量が使用されている(helpより)。
メタゲノムでお馴染みのsunburst plotとなっていて、このponge microbiomeプロジェクトの菌叢が中心の上位ランク(ドメイン/門)から外側に向かって下位ランクまでプロットされている。

サンプルのメタデータも見やすく整理されて表示される。サンプル位置を表すマップも表示されている。




コミュニティプロファイルはhelpのリンクから一括ダウンロードできる。

HPより
- Sandpiper のデータはSingleM パイプラインを用いて生成されている。このパイプラインは、NCBI SRA に登録されているパブリックメタゲノムデータセットのうち、metagenomic、またはsoil metagenomeのように「メタゲノム由来」とされる生物から得られたものに適用されている。これらのデータが基づく Genome Taxonomy Database (GTDB) のバージョンは R226 となっている。
- SingleM はショットガンメタゲノムデータからOTUの存在量を直接検出する。このツールは、35個の細菌マーカー遺伝子と37個の古細菌マーカー遺伝子(合計59遺伝子)からなる単一コピーの高保存領域を、翻訳後のアミノ酸配列としてカバーするリードを探索する。各リード中でこれら保存領域をカバーする塩基配列は、OTUとしてクラスタリングされる。重要なのは、このクラスタリングが分類学的アノテーション前に行われる点である。これにより、参照データベースへの依存度が高い手法とは一線を画している。
- SingleM では、1つの分類群に対して複数の OTU が割り当てられる場合があり、これは種内株レベルの多様性や、新奇分類群内の複数系統群を反映している可能性がある。
引用
Comprehensive taxonomic identification of microbial species in metagenomic data using SingleM and Sandpiper
Ben J. Woodcroft, Samuel T. N. Aroney, Rossen Zhao, Mitchell Cunningham, Joshua A. M. Mitchell, Rizky Nurdiansyah, Linda Blackall & Gene W. Tyson
Nature Biotechnology Brief Communication, Published: 16 July 2025