2025/05/05 追記
2025/05/06 追記
レポジトリより
このkegg-pathways-completenessというツールは、与えられたKEGGオルソログ(KO)の集合に対して、その有無に基づいて各KEGGパスウェイモジュールの完全性を計算する。このツールの現在のバージョンは、495のKEGGモジュールを持っている (2024/12/06)。
インストール
condaで環境を作ってインストールした(ubuntu22)。
mamba create -n kegg-pathways-completeness python=3.10 -y
conda activate kegg-pathways-completeness
#pip
pip install kegg-pathways-completeness
#conda
mamba install bioconda::kegg-pathways-completeness
#Docker(未テスト)
docker pull quay.io/biocontainers/kegg-pathways-completeness
> give_completeness -h
usage: give_completeness [-h] [--version] (-i INPUT_FILE | -l INPUT_LIST | -s LIST_SEPARATOR) [-g GRAPHS] [-a DEFINITIONS] [-n NAMES] [-c CLASSES] [-o OUTDIR] [-r OUTPREFIX] [-w] [-p] [-m] [-v]
Script generates modules pathways completeness by given set of KOs
options:
-h, --help show this help message and exit
--version show program's version number and exit
-i INPUT_FILE, --input INPUT_FILE
Each line = pathway
-l INPUT_LIST, --input-list INPUT_LIST
File with KOs comma separated
-s LIST_SEPARATOR, --list-separator LIST_SEPARATOR
Separator for list option (default: comma)
-g GRAPHS, --graphs GRAPHS
graphs in pickle format
-a DEFINITIONS, --definitions DEFINITIONS
All modules definitions
-n NAMES, --names NAMES
Modules names
-c CLASSES, --classes CLASSES
Modules classes
-o OUTDIR, --outdir OUTDIR
output directory
-r OUTPREFIX, --outprefix OUTPREFIX
prefix for output filename
-w, --include-weights
add weights for each KO in output
-p, --plot-pathways Create images with pathways completeness
-m, --add-per-contig Creates per-contig summary table (makes sense to use if input table has information about contigs). Does not work with --input-list option
-v, --verbose Print more logging
テストラン
hmmscanなどで得た配列とKO annotationのテキストを入力とする。
P1コンティグには3つのKO termが付与されていることを意味する。
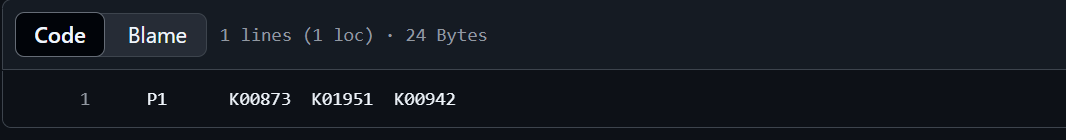
このテキストを指定して実行する。
git clone https://github.com/EBI-Metagenomics/kegg-pathways-completeness-tool.git
cd kegg-pathways-completeness-tool/tests/fixtures/give_completeness/
give_completeness -i test_pathway.txt
- -i Each line = pathway
出力例
cat summary.kegg_pathways.tsv
![]()
単純にカンマ区切りのKO listも使用できる(結果は同じ)

cd kegg-pathways-completeness-tool/tests/fixtures/give_completeness/
give_completeness -l test_kos.txt
- -l File with KOs comma separated
自分のprotein,faaで使って実行する流れ
KO termはhmmscanを使って高感度に付与することが推奨されている(link)。
#1 kofam.hmmのダウンロード
cd kegg-pathways-completeness-tool/
wget ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/pipeline-5.0/ref-dbs/db_kofam.hmm.h3f.gz
wget ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/pipeline-5.0/ref-dbs/db_kofam.hmm.h3i.gz
wget ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/pipeline-5.0/ref-dbs/db_kofam.hmm.h3m.gz
wget ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/pipeline-5.0/ref-dbs/db_kofam.hmm.h3p.gz
#2 解凍
gunzip db_kofam.hmm*
#3 hmmscanの実行(クエリはprptein)。hmmがクエリならhmmsearch
hmmscan --cpu 12 --noali --cut_ga --domtblout hmmscan_output db_kofam.hmm input_protein.faa 1> log
=> hmmscan_outputが得られる
#4 make_hmmer_table_tab_separated.pyの実行 (hmmscanのパース)(現状のスクリプトは相対インポートを使っているため、このツールの親ディレクトリに移動して以下のように実行する *1)
PYTHONPATH=. python3 -m kegg_pathways_completeness.bin.parse_hmmtable.make_hmmer_table_tab_separated -i hmmscan_output -o out.tsv
=> out.tsvが得られる
#5 parse_hmmer_tab_separated_table.pyの実行。4のoutput.tsvとprotein.faaを指定する
PYTHONPATH=. python3 -m kegg_pathways_completeness.bin.parse_hmmtable.parse_hmmer_tab_separated_table -i out.tsv -f input_protein.faa -t hmmscan
=> out.tsv_parsedが得られる
#6 最後にgive_completenessの実行 (”-i”で指定する)
give_completeness -i out.tsv_parsed -o outdir
> head out.tsv_parsed #step5の出力

可視化
mamba install -c conda-forge graphviz -y
give_completeness -i out.tsv_parsed -o outdir -p
- -p Create images with pathways completeness
outdir/pathways_plots/

その他
コメント
ゲノム1つ分のデータなら、KAASや今回のKEGG.hmmを使ったhmmscanでKOアノテーションを付けてKEGG mapperにアップロードすると結果を簡単に分析することが出来ます。
引用
GitHub - EBI-Metagenomics/kegg-pathways-completeness-tool
*1
もしくはmake_hmmer_table_tab_separated.pyの
from ..utils import __version__
を
from kegg_pathways_completeness.utils import __version__
に修正する
(Python はスクリプトを直接実行したときとパッケージの一部として実行したときでインポートのルールが変わる)。
関連