2019 6/25インストール追記, 12/17 タイトル修正
2020 7/12 help追加、タイトル修正, 7/14 helpとGitHubリンクを間違ったため差し替え, 8/11 ダウンロードリンクをカレントに修正
2022/04/07 インストール手順更新、06/24 論文引用、07/08 追記
2024/02/01 他のコマンド追記、マニュアルURL最新版に更新
HMMERはタンパク質のドメイン検索に使われるツール。Pfamなどのタンパク質ドメインのデータベースを使い、ドメインの検索を行ってアノテーションをつけることができる。ここではタンパク質配列をクエリとしてHMMプロファイルデータベースを検索するhmmscanを試す。他にも、多重配列アライメント(MSA)またはプロファイル HMM を入力とし、HMM のデータベース(PDB、Pfam、InterPro など)から相同タンパク質を検索するHHsearchなどがあある。
webサーバー
https://www.ebi.ac.uk/Tools/hmmer/
マニュアル
http://eddylab.org/software/hmmer/Userguide.pdf
hmmerブログ
https://cryptogenomicon.org/category/hmmer/
インストール
ダウンロードリンク
conda、brewで導入できる。
#bioconda (link)
mamba install -c bioconda -y hmmer
#homebrew
brew install hmmer
> hmmpress -h
$ hmmpress -h
# hmmpress :: prepare an HMM database for faster hmmscan searches
# HMMER 3.3 (Nov 2019); http://hmmer.org/
# Copyright (C) 2019 Howard Hughes Medical Institute.
# Freely distributed under the BSD open source license.
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Usage: hmmpress [-options] <hmmfile>
Options:
-h : show brief help on version and usage
-f : force: overwrite any previous pressed files
> hmmscan -h
$ hmmscan -h
# hmmscan :: search sequence(s) against a profile database
# HMMER 3.3 (Nov 2019); http://hmmer.org/
# Copyright (C) 2019 Howard Hughes Medical Institute.
# Freely distributed under the BSD open source license.
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Usage: hmmscan [-options] <hmmdb> <seqfile>
Basic options:
-h : show brief help on version and usage
Options controlling output:
-o <f> : direct output to file <f>, not stdout
--tblout <f> : save parseable table of per-sequence hits to file <f>
--domtblout <f> : save parseable table of per-domain hits to file <f>
--pfamtblout <f> : save table of hits and domains to file, in Pfam format <f>
--acc : prefer accessions over names in output
--noali : don't output alignments, so output is smaller
--notextw : unlimit ASCII text output line width
--textw <n> : set max width of ASCII text output lines [120] (n>=120)
Options controlling reporting thresholds:
-E <x> : report models <= this E-value threshold in output [10.0] (x>0)
-T <x> : report models >= this score threshold in output
--domE <x> : report domains <= this E-value threshold in output [10.0] (x>0)
--domT <x> : report domains >= this score cutoff in output
Options controlling inclusion (significance) thresholds:
--incE <x> : consider models <= this E-value threshold as significant
--incT <x> : consider models >= this score threshold as significant
--incdomE <x> : consider domains <= this E-value threshold as significant
--incdomT <x> : consider domains >= this score threshold as significant
Options for model-specific thresholding:
--cut_ga : use profile's GA gathering cutoffs to set all thresholding
--cut_nc : use profile's NC noise cutoffs to set all thresholding
--cut_tc : use profile's TC trusted cutoffs to set all thresholding
Options controlling acceleration heuristics:
--max : Turn all heuristic filters off (less speed, more power)
--F1 <x> : MSV threshold: promote hits w/ P <= F1 [0.02]
--F2 <x> : Vit threshold: promote hits w/ P <= F2 [1e-3]
--F3 <x> : Fwd threshold: promote hits w/ P <= F3 [1e-5]
--nobias : turn off composition bias filter
Other expert options:
--nonull2 : turn off biased composition score corrections
-Z <x> : set # of comparisons done, for E-value calculation
--domZ <x> : set # of significant seqs, for domain E-value calculation
--seed <n> : set RNG seed to <n> (if 0: one-time arbitrary seed) [42]
--qformat <s> : assert input <seqfile> is in format <s>: no autodetection
--cpu <n> : number of parallel CPU workers to use for multithreads [2]
実行方法
1,準備;タンパク質ドメインのデータベースを用意する必要がある。ここではPfamのデータベースを使う。
FTPサーバ Current release
ftp://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/
Pfam.version.gzをダウンロードして開く。(2017年実行)
user$ gzcat Pfam.version.gz
Pfam release : 31.0
Pfam-A families : 16712
Date : 2017-02
Based on UniProtKB : 2016_10
31.0であることが確認できる。また、公式ページの通りPfam-Aが16712登録されていることが確認できる。
このPfam-Aをデータベースに使う。Pfam-A.hmm.gzをダウンロードして解凍する。
ftpサーバー current release
ftp://ftp.ebi.ac.uk/pub/databases/Pfam/current_release
#1 ダウンロード
wget ftp://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/Pfam-A.hmm.gz
#2 解凍
gzip -dv Pfam-A.hmm.gz #解凍
#3 indexをつける(必須)
hmmpress proteins_hmm
========================================================
#補足 MSAからhmmファイルを自前で作る場合、hmmbuildコマンド&hmmpressコマンドを使う。
#1 注:MSAはStockholm alignment formatが推奨されている。esl-reformatコマンドでmuscleやmafftからのMSAを変換する
esl-reformat stockholm protein.aln > protein.sto
#2 "出力名 MSA" の順に指定
hmmbuild proteins_hmm proteins_MSA.sto
#3 続いてHMM DBを作成
hmmpress proteins_hmm
=> indexなど4つのファイルができる。ランする時にこれを指定する。
4つのファイルができる。
pfam_database]$ ls -al Pfam-A.hmm.*
-rw-rw-r-- 1 uesaka 310624148 Jul 31 10:36 Pfam-A.hmm.h3f
-rw-rw-r-- 1 uesaka 1153233 Jul 31 10:36 Pfam-A.hmm.h3i
-rw-rw-r-- 1 uesaka 568043178 Jul 31 10:36 Pfam-A.hmm.h3m
-rw-rw-r-- 1 uesaka 668124941 Jul 31 10:36 Pfam-A.hmm.h3p
========================================================
2,hmmscanのラン(ホモロジーサーチ)
hmmscan -o output.txt --cpu 1 -E 1e-10 Pfam-A.hmm input.faa
- --cpu Set the number of parallel worker threads to
- -o Direct the main human-readable output to a file
- --tblout Save a simple tabular (space-delimited) file summarizing the per-target output., with one data line per homologous target model found.
- --domtblout Save a simple tabular (space-delimited) file summarizing the per-domain output, with one data line per homologous domain detected in a query sequence for each homologous model.
hmmscanのオプション(リンク)。
終わると以下のようなアライメントファイルが出力される。

-oの代わりに--tbloutをつけると1行1クエリ形式になる。
hmmscan --domtblout output.txt --cpu 1 -E 1e-10 Pfam-A.hmm input.faa > log
#コメント削除
grep -v "#" output.txt
10タンパク質調べた例。

2022/07/08
TOP HITだけ保持する(引用)。hmmscan配列レベル出力(--tblout)はクエリごとにE-valueの低い順にプリントされる。先頭だけ取り出せばTOP HITが取り出せる。
hmmscan --tblout output --cpu 10 -E 1e-1 Pfam-A.hmm input.faa > log
awk '!x[$3]++' output > output_tophit
grep -v "#" output_tophit #コメント削除
-
--tblout save parseable table of per-sequence hits to file <f>
-
--domtblout save parseable table of per-domain hits to file <f>
output;2つのクエリ配列の結果。どちらのクエリ配列(3列目)も複数の配列がヒットしている。E-value列を見ると、どちらのクエリ配列もE-valueが低い順に並んでいる。
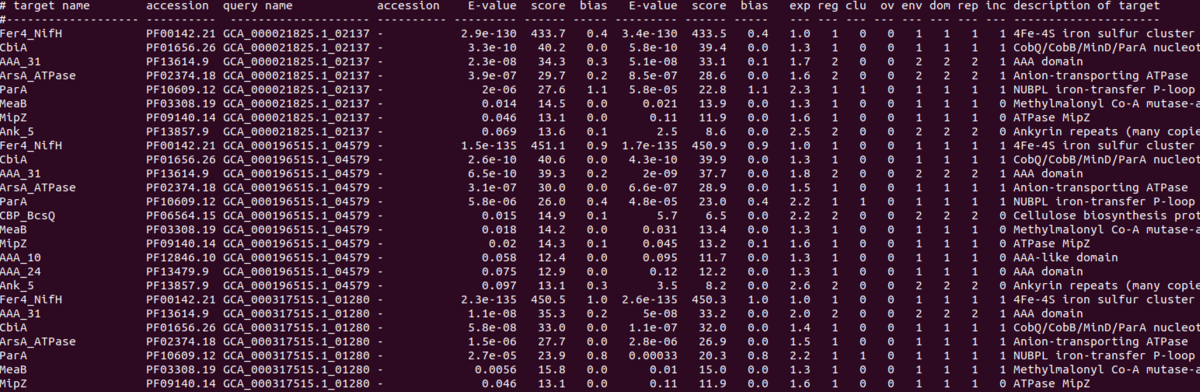
output_tophit;TOP HITがそれぞれのクエリ配列から取り出された。

他にもいくつかのコマンドがあります(wiki)。


(HMMER User’s Guideより)
引用
HMMER web server: interactive sequence similarity searching
Robert D. Finn,* Jody Clements, and Sean R. Eddy.
Nucleic Acids Res. 2011 Jul 1; 39(Web Server issue): W29–W37. Published online 2011 May 18. doi: 10.1093/nar/gkr367 PMCID: PMC3125773.
参考
HMMERを使ったPfamデータベースへのドメイン検索 - バイオインフォマティクス初心者の日常
hmmscan vs. hmmsearch speed: the numerology | Cryptogenomicon
https://www.biostars.org/p/10094/
関連
より高速
高速
https://kazumaxneo.hatenablog.com/entry/2024/02/01/235846
2022/06/24"Pfamが提供するモデルは、対象とする分類群から見て非常にgeneral(一般的)であることが多く、このような一般的なモデルでは、キングダム固有のモデルが提供するような特異性や選択性に欠ける可能性があることが以前から指摘されてた。本発表では、あるキングダムの分類群に対するHMMのドメインライブラリを作成するための一般的なアプローチを紹介する。”
(2007年の論文)
vFamsパイプライン
”RefSeqの全てのウイルス性タンパク質(非ファージ)からプロファイルHMMを構築するバイオインフォマティクスパイプラインを開発した”
(2014年の論文)