組換えは細菌において重要な進化の力であるが、ゲノムサンプルの祖先に起こった組み換えを再構築することは依然として困難である。本発表では、最尤推論を用いて細菌ゲノム中の組換えを検出し、系統樹の再構築に反映させるClonalFrameMLを紹介する。ClonalFrameMLは数百のゲノムを数時間で解析することができ、その有用性をシミュレーションと実データセットで実証した。Clostridium difficile ST6では転移因子に関連する組換えホットスポットを、Staphylococcus aureus ST582では未記載の310kb染色体置換の証拠を発見した。ClonalFrameMLは、http://clonalframeml.googlecode.com/ で自由に利用できる。
Githubより
ClonalFrameMLは細菌ゲノムの組換え推定を効率的に行うソフトウェアです。Xavier DidelotとDaniel Wilsonによって作成されました。ClonalFrameMLは、あらゆる種類のアラインメントされた配列データに適用することができますが、特に全ゲノム配列の解析を目的としています。標準的なデスクトップパソコンで、数百の全ゲノムを数時間で比較することが可能です。ClonalFrameMLの主な出力は、組換えを考慮して枝長を補正した系統樹、組換え過程の主要パラメータの推定、系統樹の各枝について組換えが起こった場所のゲノムマップの3つです。
ClonalFrameMLは、Didelot and Falush (2007)が以前に紹介したベイズソフトウェアClonalFrameを最尤法で実装したものです。ClonalFrameMLの基礎となる組換えモデルはClonalFrameと全く同じですが、この新しい実装はより高速で、より大きなゲノムデータセットを扱うことができ、MCMC収束の問題にも悩まされることはありません。
Guide
https://github.com/xavierdidelot/clonalframeml/wiki
インストール
mamba create -n clonalframeml -y
conda activate clonalframeml
mamba install -c conda-forge -c bioconda -c defaults clonalframeml -y
> ClonalFrameML
ClonalFrameML v1.12
Syntax: ClonalFrameML newick_file fasta_file output_file [OPTIONS]
Options specifying the analysis type:
-em true (default) or false Estimate parameters by a Baum-Welch expectation maximization algorithm.
-embranch true or false (default) Estimate parameters for each branch using the EM algorithm.
-rescale_no_recombination true or false (default) Rescale branch lengths for given sites with no recombination model.
-imputation_only true or false (default) Perform only ancestral state reconstruction and imputation.
Options affecting all analyses:
-kappa value > 0 (default 2.0) Relative rate of transitions vs transversions in substitution model
-fasta_file_list true or false (default) Take fasta_file to be a white-space separated file list.
-xmfa_file true or false (default) Take fasta_file to be an XMFA file.
-ignore_user_sites sites_file Ignore sites listed in whitespace-separated sites_file.
-ignore_incomplete_sites true or false (default) Ignore sites with any ambiguous bases.
-use_incompatible_sites true (default) or false Use homoplasious and multiallelic sites to correct branch lengths.
-show_progress true or false (default) Output the progress of the maximum likelihood routines.
-chromosome_name name, eg "chr" Output importation status file in BED format using given chromosome name.
-min_branch_length value > 0 (default 1e-7) Minimum branch length.
-reconstruct_invariant_sites true or false (default) Reconstruct the ancestral states at invariant sites.
-label_uncorrected_tree true or false (default) Regurgitate the uncorrected Newick tree with internal nodes labelled.
Options affecting -em and -embranch:
-prior_mean df "0.1 0.001 0.1 0.0001" Prior mean for R/theta, 1/delta, nu and M.
-prior_sd df "0.1 0.001 0.1 0.0001" Prior standard deviation for R/theta, 1/delta, nu and M.
-initial_values default "0.1 0.001 0.05" Initial values for R/theta, 1/delta and nu.
-guess_initial_m true (default) or false Initialize M and nu jointly in the EM algorithms.
-emsim value >= 0 (default 0) Number of simulations to estimate uncertainty in the EM results.
-embranch_dispersion value > 0 (default .01) Dispersion in parameters among branches in the -embranch model.
-output_filtered true of false (default) Output a filtered alignment including only non-recombinant sites.
Options affecting -rescale_no_recombination:
-brent_tolerance tolerance (default .001) Set the tolerance of the Brent routine for -rescale_no_recombination.
-powell_tolerance tolerance (default .001) Set the tolerance of the Powell routine for -rescale_no_recombination.
実行方法
ClonalFrameMLの実行には、2つの入力ファイルが必要。1つはNewickフォーマットで作成されたツリーファイルで、もう1つは、配列のアラインメントファイルで、FastaフォーマットかExtended Multi Fasta (XMFA)フォーマットの必要がある。ツリーにはアラインメントファイルの配列と同じ数のleaveが必要であり、leave名はアラインメントファイルのヘッダーの名前と一致しなければならない。
ClonalFrameML newick_tree_file sequence.aln output_prefix
出力
14genomeの系統樹ファイルの計算には2時間ほどかかった( TR3990x)。
出力ファイルについて(Githubより)
ML_sequence.fasta
このファイルには、系統樹のすべての内部ノードと、入力配列のすべての欠損データに対して最尤法で再構築された配列が含まれている。系統樹の各葉と各内部ノードに対する配列を持つアライメントである。このアラインメントでは、入力ファイル中の配列のユニークなパターンごとに1つの位置が存在する。入力ファイルとML_sequence.fasta出力ファイルの位置の対応は、position_cross_reference.txt出力ファイル(下記参照)に記載されている。
position_cross_reference.txt
入力配列ファイル中の各位置と、出力ファイルであるML_sequence.fasta中の配列の対応する位置をカンマ区切りで示したベクトル。position_cross_reference.txtの値は、ML_Sequence.fastaファイルに沿った位置に対応する。例えば、入力ファイルの10番目の位置で何が再構成されたかを調べるには、まず position_cross_reference.txt で10番目の値を調べ、これが例えば5に等しければ、ML_sequence.fasta ファイル中の5番目の位置を調べる必要がある。
em.txt
このファイルには、R/θ、ν、δ、枝長の点推定値が含まれている。
emsim.txt
このファイルには3つのパラメータR/theta, nu, deltaのブートストラップ値が含まれている。
importation_status.txt
このファイルには、再構成された組換えイベントのリストが含まれている。各イベントごとに1行あり、1列目はイベントが見つかった枝を示し、2列目と3列目は組換えイベントの影響を受けた最初と最後のゲノム位置を示している。
labelled_tree.newick
このファイルには、他のファイルで参照できるように、すべてのノードにラベルを付けた出力ツリーが含まれている。
視覚化
#Rが使える環境で実行
#依存
mamba install -c bioconda r-phangorn -y
#本体
git clone https://github.com/xavierdidelot/ClonalFrameML.git
Rscript ClonalFrameML/src/cfml_results.R output_prefix
出力例
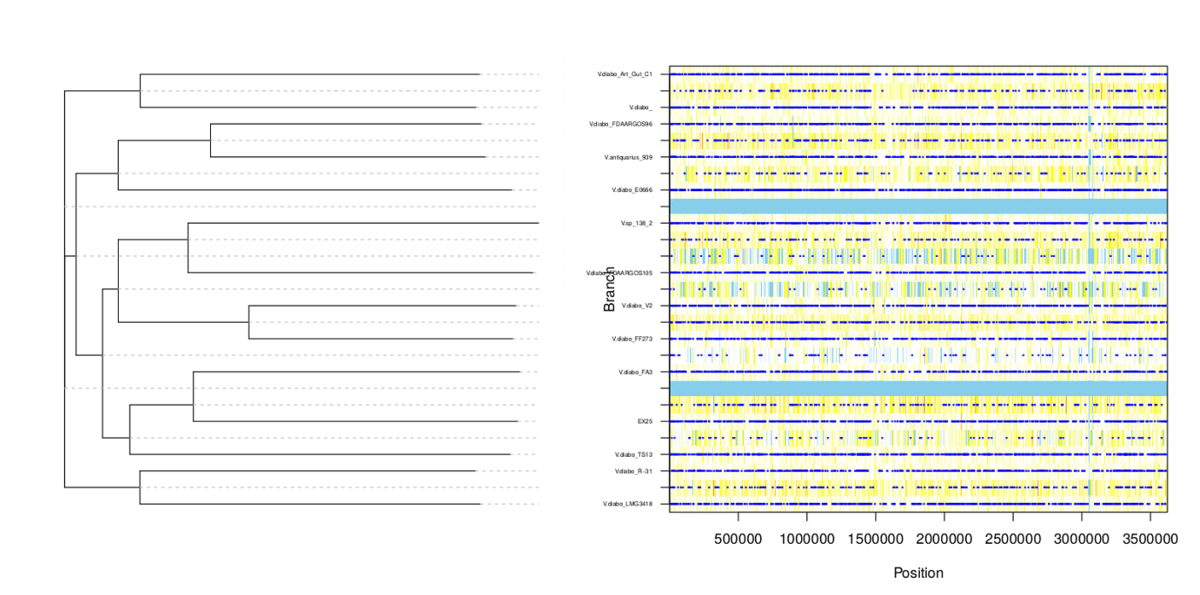
内部ノードと枝の先端(葉)部分について表示される。青点が組み換えイベントを表す。
Guideには実践的な例が用意されています。使用前に読まれることを推奨します。
引用
ClonalFrameML: efficient inference of recombination in whole bacterial genomes
Xavier Didelot, Daniel J Wilson
PLoS Comput Biol. 2015 Feb 12;11(2)