RNA-Seq実験で得られたコンティグを含め、過去10年間に蓄積された配列情報は飛躍的に増加しており、リードデータをアセンブルする際にはキメラ配列の定量が必須となっている。トランスクリプトームでは、de novoでアセンブリされたキメラは、基本的な転写産物によく似ているが、共進化部位間に見られるパターンや、マッピングされたリードカウントなどは不明瞭になる。本著者らは、RNA-Seqデータ用のde Bruijnベースのde novo assemblerを開発した。このアセンブラは、コンティグが作成される基本的なグラフの複雑さを表す分類システムを利用している。各コンティグは、曖昧なパスが存在するかどうかを示す3つのレベルのうちの1つでラベル付けされる。この副産物として、基礎となる遺伝子ファミリーの複雑さの範囲に関する情報が得られる。CStonesが高品質のコンティグをアセンブリし、このようにラベル付けする能力を実証するために、シミュレートされたデータと実際のデータの両方を使用した。模擬データでは、Drosophila melanogaster、Panthera pardus、Rattus norvegicus、Selinus canariaの4種のcDNAライブラリから1,000万のリードペアを生成した。これらのライブラリは、CStone、Trinity、rnaSPAdesを用いてアセンブルした。実データとしては、2つの成体D. melanogasterの全身サンプルを対象とした、それぞれが約3,000万のリードペアからなる2つのRNA-Seqデータセットを使用した。CStoneが作成したコンティグは、長さ、アラインメントされた領域の配列の同一性、表現されたcDNA転写物の範囲の点で、TrinityやrnaSPAdesのコンティグと同等の品質であり、同時にキメラに関する追加情報を提供した。ここでは、CStonesのアセンブリと分類プロセスの詳細を説明し、同様の分類システムを他のde novoアセンブリツールに組み込むことができることを提案する。また、関連する副次的研究として、リファレンスセット内のキメラが発現量の異なる遺伝子の同定に与える影響を調べている。CStoneは、https://sourceforge.net/projects/cstone/から入手できる。
インストール
CStone.jarファイル、ライセンス、クイック・スタート・ガイドを含むZIPファイルをsourceforgeからダウンロードしてテストした(ubuntu18)。
依存
cstoneはUbuntu 20.04、Windows 10、MacOS High Sierraでテストされている。 Cstoneは、次世代配列データの解析に適したスペックのサーバー上で動作するように設計されている。例えば、Cstoneがテストされたマシンは、32コアと128GBのメモリ、24コアと64GBのメモリを搭載している。
> dragen-os --help
$ java -jar cstone.jar
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
* * * *
* * Title: CStone v0.01 * *
* * Website: https://sourceforge.net/projects/cstone/ * *
* * License: GNU General Public License * *
* * Funder: https://www.fct.pt * *
* * * *
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
* * * *
* * No output directory (-o) specified. * *
* * * *
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
* * * *
* * * *
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
マニュアルではソースコードからIDE上でビルドする手順も簡単に説明されています。
テストラン
レポジトリで公開されているテストデータ(シミュレーションデータ、ペアエンド)をランする。
java -jar -Xmx64G cstone.jar -r1 r1_test_data.fq.gz -r2 r2_test_data.fq.gz -gz y -p 32 -o outdir
- -r1 (required) path to the forward reads (in fastq or compressed fastq format).
- -r2 (optional) path to the reverse reads (in fastq or compressed fastq format).
- -gz (default = n) indicates if the reads are compressed or not .gz (y=yes / n=no).
- -p (default = 24) indicates the number of cores required.
- -o (required) path to output directory.
- -k indicates kmer length. Default is 40. Max. value is: 48, min value is: 24
出力例
outdir/

contigs.fastaにはアセンブルされた転写産物を含まれている。各配列のタイトルにはグラフ分類が表示されている。これは、コンティグがnon-chimericであることを保証できるかどうか示されている。
$ grep ">" contigs.fasta |head -n 20

整数IDと、作成元のグラフ分類レベルのタイトルが付けられる(下も参照)。
classificationsummary.txt: 7つの分類カテゴリーのそれぞれに属するコンティグの数をまとめたもの。
$ cat classificationsummary.txt
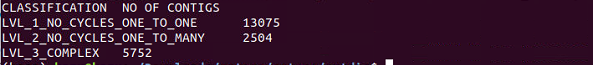
このテストデータでは、グラフのパスから配列は3つの分類レベル1-3に分けられている。詳細は論文の図3A-Cとその説明を参照(link)。
- すべてのグラフから「実行可能な可能性のある」パスをすべて出力することは不可能であるため、グラフごとにコンティグ数を指定する必要がある。グラフによっては、数十から数百のパスが存在する可能性があるため。著者らは、コンティグの非キメラ性についての洞察を得ることができる最大カバレッジに基づいて、上位3つの最良のパスを出力することを選択した(デフォルト設定)
引用
CStone: A de novo transcriptome assembler for short-read data that identifies non-chimeric contigs based on underlying graph structure
Raquel Linheiro, John Archer
PLoS Comput Biol. 2021 Nov 23;17(11):e1009631
関連