2023/04/10 タイトル変更
2023/04/11追記
2024/04/20 dockerイメージ追記
2025/01/19 分かりにくい文章を修正
純粋培養やメタゲノムから得られる微生物のゲノム配列の増加は、全ゲノムおよびショットガンシーケンス法の現在の達成可能性を反映している。しかし、ゲノムの可視化のためのソフトウェアは、自動化、異なる解析の統合、経験の浅いユーザー向けのカスタマイズ可能なオプションがまだ不十分である。本研究では、微生物ゲノムと配列エレメントの解析と可視化のために、カスタム環状ゲノム表現のプロットを作成できるPythonコマンドラインツールであるGenoViを紹介する。25種類のカラーパレット(5種類の色盲用セーフパレットを含む)、テキストフォーマットオプション、完全ゲノムや複数のレプリコン/配列を持つ配列エレメントの自動スケーリングなどのカスタマイズ可能なオプションを備えており、完全長ゲノムやドラフトゲノムを扱うように設計されている。GenoVi は、Genbank 形式ファイルを入力ファイルとして、またはディレクトリ内の複数のファイルを使用して、(i) GenBank アノテーションファイルからゲノムの特徴を可視化し、(ii) DeepNOG による Cluster of Orthologs Group (COG) カテゴリ分析を統合して、(iii) 完全長ゲノムまたは複数の配列エレメントの各レプリコンの可視化を自動的に拡大し、 (iv) COG ヒストグラム、COG スペクトラムヒートマップと各レプリコンまたはコンティグの処理の一般統計値を含む出力テーブルを生成する。GenoViの可能性は、細菌と古細菌のシングルゲノムとマルチゲノムを解析することで評価された。Paraburkholderiaのゲノムは、大規模なマルチパーティットゲノムにおけるレプリコンの高速分類を得るために解析された。GenoViは、使いやすいコマンドラインツールとして動作し、カスタマイズ可能なオプションにより、科学論文、教育リソース、アウトリーチ活動用のゲノムマップを自動生成する。GenoViは自由に利用でき、https://github.com/robotoD/GenoVi からダウンロードできる。
I'm very excited to introduce you to GenoVi. An easy-to-use tool for prokaryotic Genome Visualization.
— Andrés Cumsille (@a_cumsille) April 5, 2023
It's been Just published in @PLOSCompBiol.
Excellent work in collab with @AgRoboto, Andrea Rodriguez, and Vicente Saona
#PLOSCompBio @usantamariahttps://t.co/EIVO7n4dpK
User guide
User guide · robotoD/GenoVi Wiki · GitHub
インストール
レポジトリの説明に従って、condaでpython3.7の環境を作ってpipで導入した(intel mac miniとubuntu使用)。
依存
- Circos 0.69-8
- Python 3.7 or later
- DeepNog 1.2.3
- NumPy 1.20.2
- MatPlotLib 3.5.2
- Pandas 1.2.4
- Biopython 1.79
- CairoSVG 2.5.2
- Seaborn 0.12
- Perl 5
- List::MoreUtils (Perl library)
- Natsort 8.2.0
mamba create -n genovi python=3.7 circos -y
conda activate genovi
pip install genovi
#circosが入らなかったので改めて導入
mamba install -c bioconda circos
#docker(非公式?)
docker pull staphb/genovi:latest
#run
docker run --rm -itv $PWD:/data/ -w /data staphb/genovi:latest bash
> genovi -h
> genovi -h
$ genovi -h
usage: genovi [-h] -i INPUT_FILE -s {complete,draft} [-o OUTPUT_FILE] [-cu] [-b DEEPNOG_CONFIDENCE_THRESHOLD] [--cogs COGS] [-a {center,top,bottom,A,<,U,matrix,two_lines}]
[--scale {variable,linear,sqrt}] [-k] [-r] [-w WINDOW] [-v VERBOSE] [-c] [-cp {top,bottom,bottom-right,auto}] [-t TITLE] [--title_position {center,top,bottom}] [--italic_words ITALIC_WORDS]
[--size] [-te] [-cs COLOUR_SCHEME] [-bc BACKGROUND] [-fc FONT_COLOUR] [-pc CDS_POSITIVE_COLOUR] [-nc CDS_NEGATIVE_COLOUR] [-tc TRNA_COLOUR] [-rc RRNA_COLOUR] [-cc GC_CONTENT_COLOUR]
[-sc GC_SKEW_COLOUR] [-sl GC_SKEW_LINE_COLOUR] [--version]
optional arguments:
-h, --help show this help message and exit
-i INPUT_FILE, --input_file INPUT_FILE
Genbank file (.gb, .gbk, .gbff, .gbff.gz) path
-s {complete,draft}, --status {complete,draft}
To draw each sequence as a unique circular representation (complete) or as a circle with bands per sequence (draft).
-o OUTPUT_FILE, --output_file OUTPUT_FILE
Directory for output files. Default: genovi
-cu, --cogs_unclassified
Do not classify each protein sequence into COG categories.
-b DEEPNOG_CONFIDENCE_THRESHOLD, --deepnog_confidence_threshold DEEPNOG_CONFIDENCE_THRESHOLD
Lower threshold for DeepNOG prediction certainty to be considered. Values in range [0,1] Default: 0
--cogs COGS Symbol of each COG to draw. For example, for drawing only information storage and processing related, use 'ABJKLX'. When using a number n, the n most common categories will be
drawn. By default, draws all of them.
-a {center,top,bottom,A,<,U,matrix,two_lines}, --alignment {center,top,bottom,A,<,U,matrix,two_lines}
When using --status complete, this defines the vertical alignment of every circular representation. Options: center, top, bottom, A (First on top), < (first to the left), U (Two
on top, the rest below), matrix (multiple rows). By default this is defined by contig sizes
--scale {variable,linear,sqrt}
To select the scale-up ratio between each circular representations when the file is processes as a complete genome. This is useful to ensure visibility of each representation when
the length difference is too high. Options: variable, linear, sqrt. Default: sqrt
-k, --keep_temporary_files
Do not delete files used for circos image generation, including protein categories prediction by Deepnog.
-r, --reuse_predictions
If available, reuse DeepNog prediction result from previous run. Useful only after --keep_temporary_files flag enabled.
-w WINDOW, --window WINDOW, --step WINDOW
Base pair window for GC plotting. Default: 5000
-v VERBOSE, --verbose VERBOSE
Weather to print progress log.
--version show program's version number and exit
text:
-c, --captions_not_included
Do not include captions for genomic features and COG colors.
-cp {top,bottom,bottom-right,auto}, --captions_position {top,bottom,bottom-right,auto}
Where to position figure captions. Options: top, bottom, bottom-right.
-t TITLE, --title TITLE
Title of the image (e.g., strain taxomomical identification). By default, it does not include title
--title_position {center,top,bottom}
--italic_words ITALIC_WORDS
How many of words of the title should be written in italics. WARNING: Italics is not supported by the svg-to-png engine, so PNG image will not be available if this option is used.
Default: 0
--size To write the size (in base pairs) inside each circle.
-te, --tracks_explain
To include an additional text on each track to specify the meaning for each one.
colours:
-cs COLOUR_SCHEME, --colour_scheme COLOUR_SCHEME, --colour COLOUR_SCHEME
Colour scheme to use. Individual colours may be overriden wih other arguments. COG colours can not be edited. Options: neutral, blue, purple, soil, grayscale, velvet, pastel,
ocean, wood, beach, desert, ice, island, forest, toxic, fire, spring
-bc BACKGROUND, --background BACKGROUND, --background_colour BACKGROUND
Background colour. Default: transparent
-fc FONT_COLOUR, --font_colour FONT_COLOUR
Colour for ticks and caption texts. Default: black
-pc CDS_POSITIVE_COLOUR, --CDS_positive_colour CDS_POSITIVE_COLOUR
Colour for positive CDSs, in R, G, B format. Default: '180, 205, 222'
-nc CDS_NEGATIVE_COLOUR, --CDS_negative_colour CDS_NEGATIVE_COLOUR
Colour for negative CDSs, in R, G, B format. Default: '53, 176, 42'
-tc TRNA_COLOUR, --tRNA_colour TRNA_COLOUR
Colour for tRNAs, in R, G, B format. Default: '150, 5, 50'
-rc RRNA_COLOUR, --rRNA_colour RRNA_COLOUR
Colour for rRNAs, in R, G, B format. Default: '150, 150, 50'
-cc GC_CONTENT_COLOUR, --GC_content_colour GC_CONTENT_COLOUR
Colour for GC content, in R, G, B format. Default: '23, 0, 115'
-sc GC_SKEW_COLOUR, --GC_skew_colour GC_SKEW_COLOUR
Colour scheme for GC skew. Might be a pair of RGB colours or Circos-readable code. For details, please read CIRCOS documentation. Default: '140, 150, 198 - 158, 188, 218'
-sl GC_SKEW_LINE_COLOUR, --GC_skew_line_colour GC_SKEW_LINE_COLOUR
Colour for GC skew line. Default: black
実行方法
1,ドラフトゲノム
"-s draft"の場合(デフォルト)、全コンティグがまとめて1つ環状ゲノムでプロットされる。
genbankファイル(.gbkか.gbff、gzip形式も認識)とゲノムのステータス(完全長かドラフトのどちらか)を指定する。-csでカラースキームを、-bcで背景色を指定できる(デフォルトはなし)。ここではチュートリアルのデータを指定。
git clone https://github.com/robotoD/GenoVi.git
cd GenoVi/
genovi -i input_test/Corynebacterium_alimapuense_VA37.gbk -s draft -cs paradise -cu -bc white -o genovi
- -i GenBank input file path.
- -o Output file name. Default: genovi.
- -s “complete” or “draft” Complete genomes are drawn as separate circles for each contig/replicon.
- -cs Prebuilt color scheme to use for CDS, RNAs, and GC analysis. Options: strong,autumn,dawn,blossom,paradise,neutral, blue, purple, soil, grayscale, velvet, pastel, ocean, wood, beach, desert, ice, island, forest, toxic, fire, spring.
- -cu Do not classify each coding sequence into Clusters of Orthologous Groups of proteins (COGs).
- -bc Background colour, in R, G, B format. By default, it has no background.
出力
genovi/
genovi.png
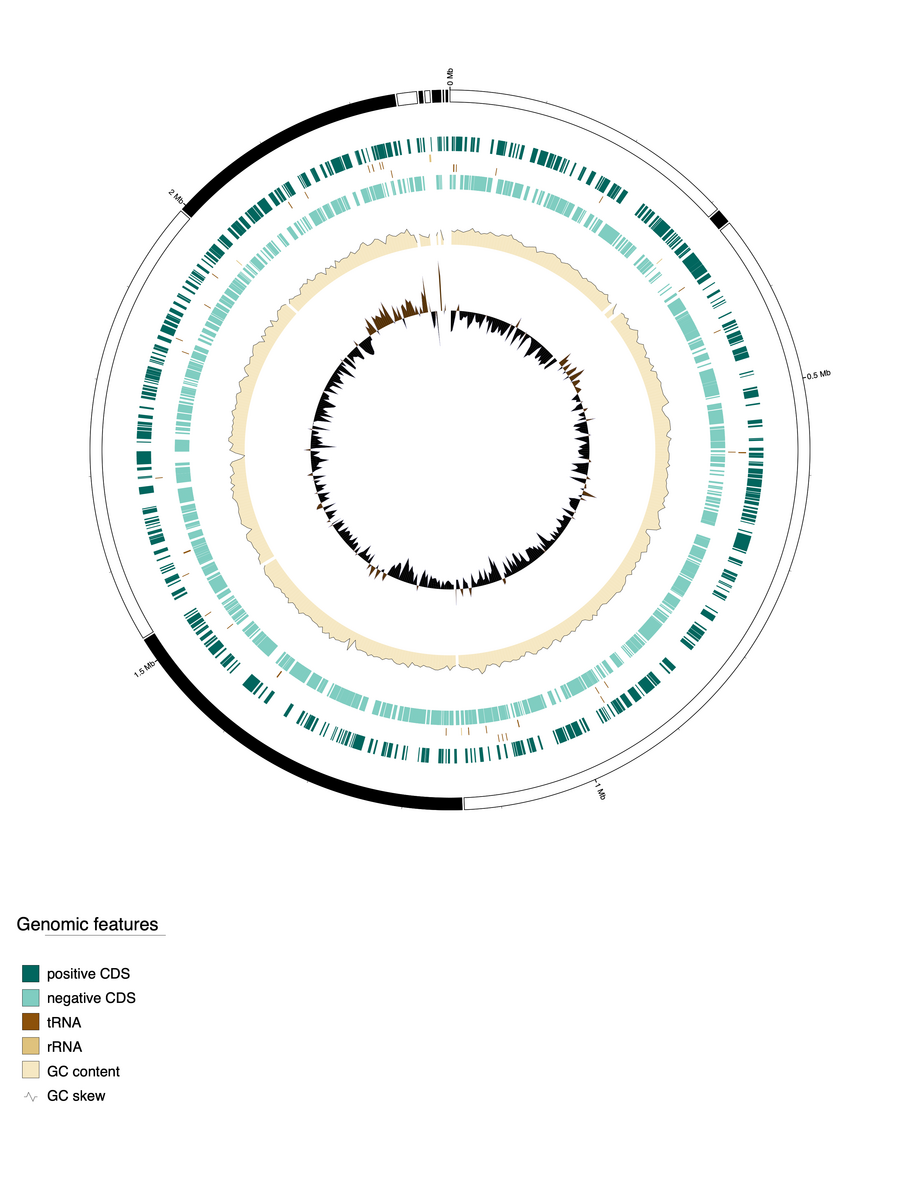
Corynebacterium alimapuense VA37のドラフトゲノムのすべてのコンティグが1つの円中に描かれている。外側から内側に向かって、コンティグ長(各コンティグは黒と白で交互に描かれている)、プラス鎖とマイナス鎖のコーディング配列(CDS)、GCコンテンツ、そして最後にGCスキューが表示される。
デフォルトでは、DeepNOGを使って各CDSのCOG(Clusters of Orthologous Groups of proteins)が予測される。”-cu”フラグを使用すると、CDSをCOGに分類しないように指定できる。これにより、DeepNOGがマシンにインストールされていない場合でもプログラムを実行できる。デフォルトでは、すべてのCDSのCOG分類もプロットされる。"--cogs"を指定すると特定のCOGカテゴリだけ表示できる(レポジトリの表参照)。
2,完全長ゲノム
"-s complete"を付ける。completeを選択した場合、1つ配列ごとに環状マップにプロットされる。
genovi -i input_test/Acinetobacter_radioresistens_DD78.gbff -cs strong -s complete --size -bc white
- --size To display the genome size (in base pairs) of each circular representation.
染色体やプラスミドごとに別名で1.pngと1.svg、2.pngと2.svgなどの画像ファイルとして生成される。最後に統合された図が描画される。各配列のプロットのサイズは平方根のスケールで拡大縮小されて表示される(リニアスケールが必要な場合は”-scale linear”を使用)。
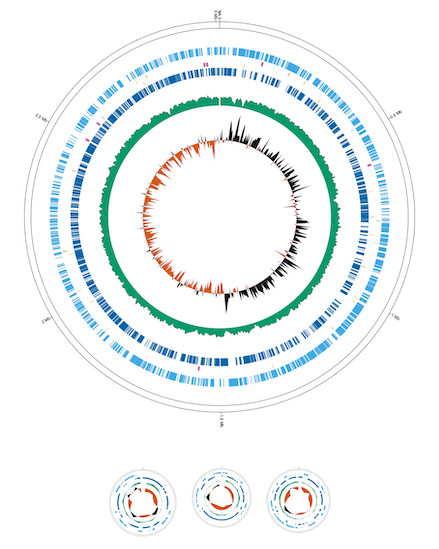

ゲノムの順番を変更することも可能
3,COGカテゴリのプロット
-cuなしだと、COGカテゴリへのアサインが行われ、COGもプロットされる(最初のコマンドから-cuのみ外した)。計算は少し時間がかかる。
genovi -i input_test/Corynebacterium_alimapuense_VA37.gbk -s draft -cs paradise -bc white -o genovi
genovi/

COG カテゴリを表示するヒストグラム、各レプリコンの COG 分類を表示するファイル、 各レプリコンのサイズ、GC含有量、CDS数、tRNA、rRNAなどの一般情報を表示するcsvファイル、各レプリコン内のCOGの分布を表示するヒートマップも含まれる。
図にはCOGのトラックと凡例が追加される。

各COGのbar plot

4,複数ゲノム
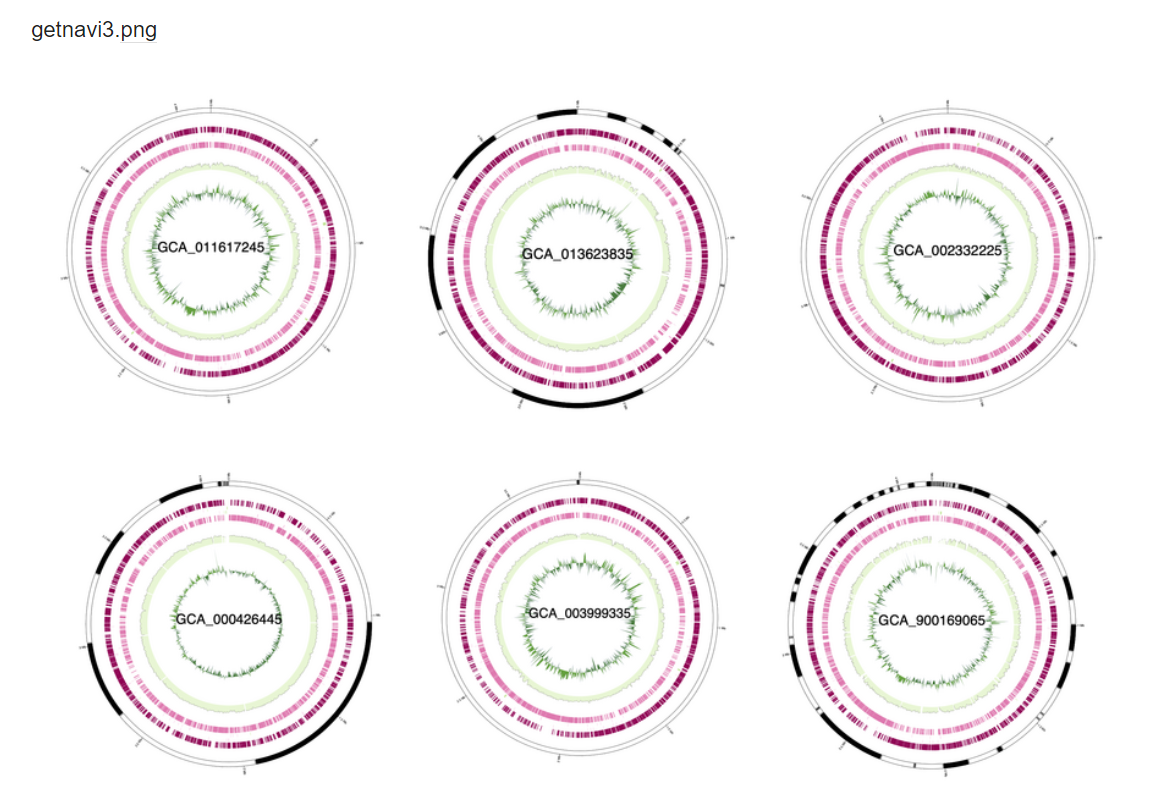
ディレクトリを入力として複数のゲノムを一度に描画することもできる。-titleをつけるとファイル名で環状プロットの中央に名前が付けられる。ディレクトリを指定した場合、その中にあるすべてのファイルが描画され、サマリーテーブルも生成される。 deepCOGなど計算時間がそれなりにかかるので注意する。
genovi -i input_test/Brevibacterium_Genomes -cs blossom -s draft --title 'filename' -bc white
- --title Figures title, for example, which genome is being represented.
出力として、各ファイルの環状表現、一般統計、COGs情報を含むフォルダが保存される。さらに、全ゲノムの一般統計情報、COG識別情報、COG頻度を1つのファイルにまとめた出力表と、すべてのゲノムを統合した図が保存される。
getnavi3/

その他
- -kまたは--keep_temporary_filesを指定するとユーザーガイドの引数で説明されているファイルも保存される。
- Intel mac miniでは、-cuをつけてCOGのプロットも表示するとエラーになった。ubuntu18LTSでは問題なく動作した。
引用
GenoVi, an open-source automated circular genome visualizer for bacteria and archaea
Andrés Cumsille, Roberto E Durán, Andrea Rodríguez-Delherbe, Vicente Saona-Urmeneta, Beatriz Cámara, Michael Seeger, Mauricio Araya, Nicolás Jara, Carlos Buil-Aranda
PLoS Comput Biol. 2023 Apr 4;19(4)
関連