品質管理はNGSパイプラインの基本的なステップであるが、軽視されがちである。クロスサンプルコンタミネーションやサンプルスワップのような問題を検出することは、データの完全性を管理するために不可欠である。ここでは、ヒトの全ゲノムおよび全トランスクリプトームシーケンスデータにおけるクロスサンプル混入、サンプルの入れ替え、および報告されたものと推定された遺伝学的およびトランスクリプトーム的性別間のミスマッチを検出する新しいPythonツールであるNGSTroubleFinderを紹介する。NGSTroubleFinderはPythonで実装され、Cで書かれたカスタムビルドの並列化パイルアップエンジンを組み込んでいる。このツールは、結果の解釈を容易にするためのキープロットを含む、サンプルに関する広範な情報をテキスト形式とHTML形式の両方で報告する。
NGSTroubleFinderはPythonとCで書かれており、pipで簡単にインストールできる。ツールのソースコードとモデルはgithub (https://github.com/STALICLA-RnD/NGSTroubleFinder)で自由に利用でき、コンテナ化されたバージョンはdockerhub (https://hub.docker.com/r/staliclarnd/ngstroublefinder)で利用できる。
NGSTroubleFinderは以下について分析する:
インストール
The project have been tested with
- python==3.10.10
- matplotlib==3.8.2
- numpy==1.26.0
- pandas==2.2.0
- onnxruntime==1.20.1
- scipy==1.12.0
- plotly==5.24.1
- seaborn==0.13.2
The package also requires libhts-dev for the C module.
sudo apt update
sudo apt install -y --no-install-recommends libhts-dev
#pip
git clone https://github.com/STALICLA-RnD/NGSTroubleFinder.git
cd NGSTroubleFinder/
python3 -m pip install .
#conda
git clone https://github.com/STALICLA-RnD/NGSTroubleFinder.git
cd NGSTroubleFinder/
conda install bioconda::htslib
python3 -m pip install .
#docker(link)
docker pull staliclarnd/ngstroublefinder
> ngsTroubleFinder -h
usage: NGS Trouble Finder [-h] -m METADATA -o OUTPUT [-t TRANSCRIPTOMIC] [-p PROCESSES] [-r REFERENCE]
NGS Trouble Finder A tool for detection and quantification of contamination and kinship across NGS data
options:
-h, --help show this help message and exit
-m METADATA, --metadata METADATA
Path to the cohort metadata. The file must be a tsv with Sample_Name, Bam_Path, Sequencing and Sex fields. The sequencing must be DNA (WGS) or RNA (rna-seq). Sex must be Male or Female.
-o OUTPUT, --output OUTPUT
Path to the output folder
-t TRANSCRIPTOMIC, --transcriptomic TRANSCRIPTOMIC
Path of the salmon count file. Sample Name must match the one provided in the metadata.
-p PROCESSES, --processes PROCESSES
Number of processes used by the pileup engines [Default: 4]
-r REFERENCE, --reference REFERENCE
Path to the reference genome fasta. Used only for CRAMS
実行方法
cramの形式のアラインメントとゲノム(bam指定時は不要)、メタデータが必須となっている。任意でsalmonのカウントマトリックスを使用する(RNA seqデータ使用時)。
タブ区切りのメタデータファイル.tsvを用意する必要がある。サンプル名、cramファイルのパス、シークエンスの種類、サンプルの性別を記載する。1行目は以下の形式である必要がある。

テストラン
レポジトリに記載されたURLからリファレンスゲノムとcram2つ :NA12891、NA12878をダウンロードすることができる。上記のメタデータを用意してランする。
#ここではイメージを立ち上げてからラン
docker run --rm -itv $PWD:/tmp/test --rm -w /tmp/test staliclarnd/ngstroublefinder:1.0.3 bash
> ngsTroubleFinder -m metadata.tsv -o out -r reference.fa
出力例
out/
- report.html: ツールからの全情報とプロットを含むインタラクティブな出力
- qcReport.tsv: サンプルごとの収集済み全情報を含む機械可読出力
- images: 全プロットをpngおよびsvg形式で含むフォルダ
- pileups: 全サンプルのパイルアップを含むフォルダ
- haplotypes: 全サンプルのハプロタイプ情報を含むフォルダ
relatedness: 血縁関係表の統計情報を含むフォルダ
report.html
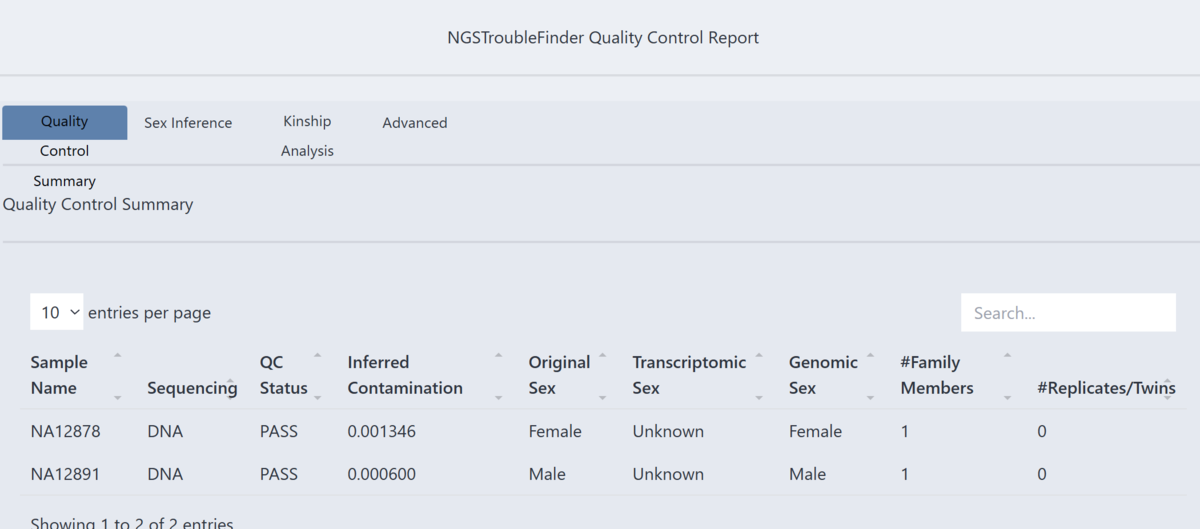
QCステータスがPASSとなっており、どちらのサンプルも品質上の問題は検出されていない。推定コンタミネーション率(Inferred Contamination)が非常に低く、実質的にコンタミネーションがないと判断できる。
Sex inference
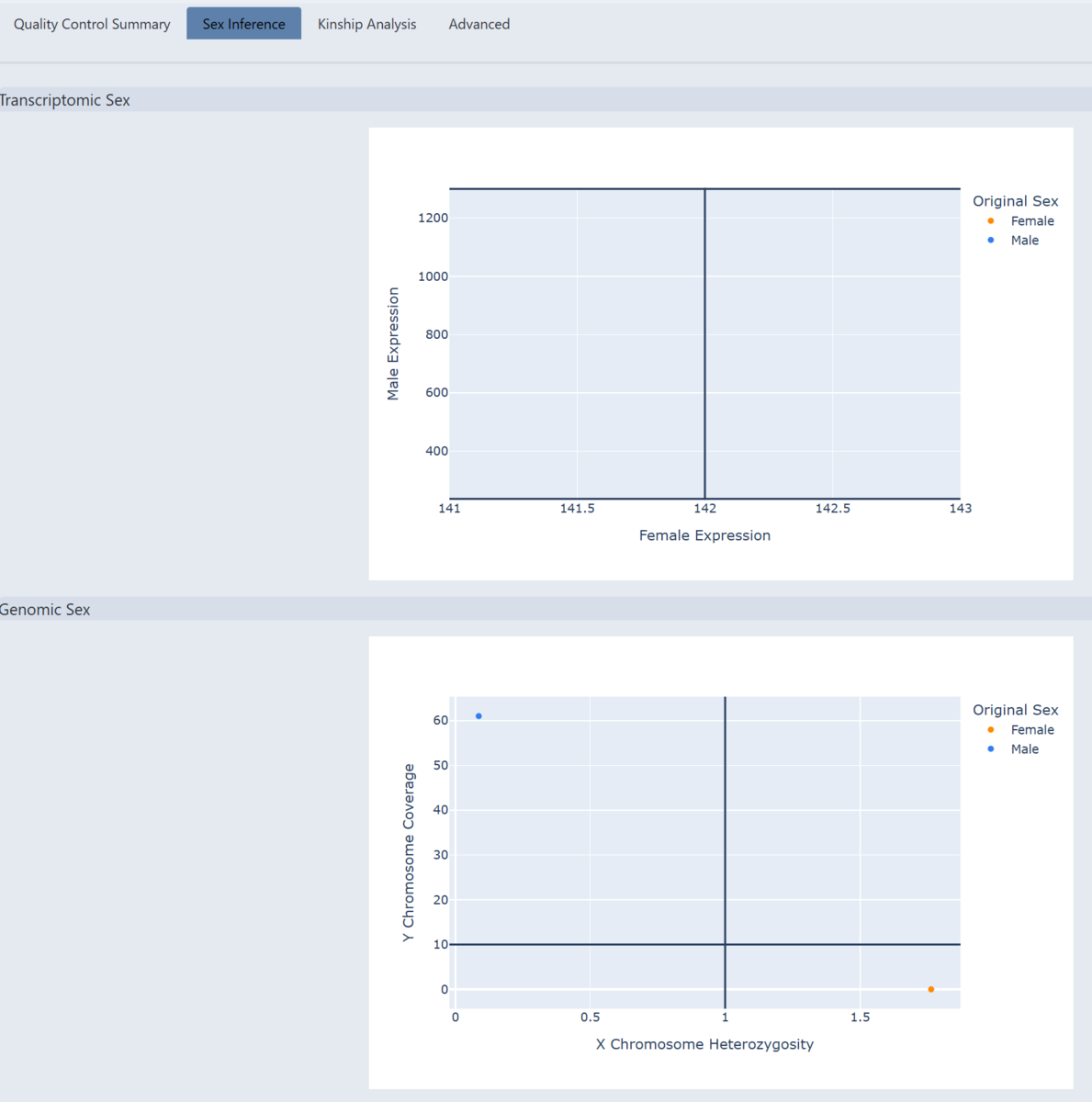
上はRNA seqデータを提供していないのでプロットなし、下のGenomic Sexはわかりにくいが2つプロットがある。この図はX染色体ヘテロ接合率とY染色体カバレッジの2軸でプロットされていて、NA12891(Male)はYカバレッジが高く、Xヘテロ接合率が低い位置に点があるため、男性と正しく判定されている。NA12878(Female)はYカバレッジが0付近で、Xヘテロ接合率が高い位置に点があるため、女性と正しく判定されている。
Kinship Analysis (wiki)
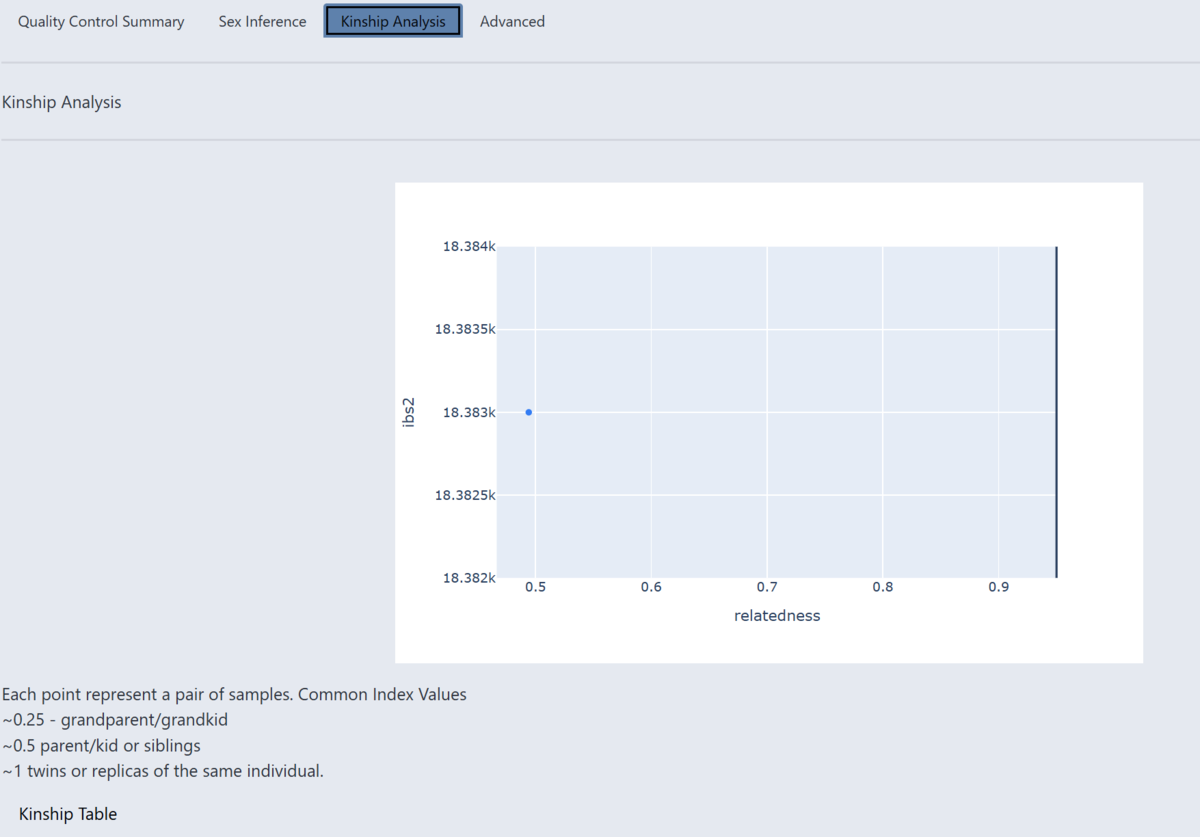
各点はサンプルのペアを表す。Y軸はibs2で、これは2つの個体が 両方のアリルを同一 として持つ SNP の数を表す。図では18383となっている。X軸はRelatednessで、これは遺伝的な類似度(血縁関係の近さ)を示す係数となっている。このペアでは0.5 に近い。
Relatednessの一般的な指標(図の説明より)
- ~0.25 - 祖父母/孫
- ~0.5 - 親子または兄弟姉妹
- ~1 - 双子または同一個人の複製
ibs2 が高いことから、遺伝的に非常に近いことが示唆される。さらにRelatednessが0.5 に近いことから、この2検体は親子または兄弟か姉妹に相当するという結果となる。
Advanced

(empty)
論文より
- NGSTroubleFinderはWGSおよびWhole-Transcriptomic RNA Sequencing(WTS)データの両方において、サンプル間コンタミネーションおよびサンプル取り違えを検出・定量化する。NGSTroubleFinderは、一般的なバリアント情報を活用した単純だが強力な線形回帰モデルに基づいており、交差コンタミネーションの定量化だけでなく、サンプル間の取り違えや血縁関係に関する洞察も提供する。さらに、サンプル汚染および血縁関係推定に加えて、NGSTroubleFinderは、バリアントおよびトランスクリプトーム情報に基づいてNGSデータから推定される生物学的性とメタデータに記載された性が一致しない性不一致も特定する。
- NGSTroubleFinderは、一般集団におけるMAF(minor allele frequency:対立遺伝子頻度)が0.1から0.9の範囲にある非常に一般的なヒトバリアントの精選セットに基づいている。二倍体アリルバリアントはdbSNP(build id 151)(Sherry 2001)から抽出されている。この精選バリアントセットはツールに同梱されており、GitHubリポジトリから入手できる。
引用
NGSTroubleFinder: A tool for detection and quantification of contamination and kinship across human NGS data
Samuel Valentini, Tecla Venturelli, Xavier Gallego, Laura Perez-Cano, Emre Guney
bioRxiv, Posted February 05, 2025.
関連