ハイスループットシーケンス技術(Next Generation Sequencing; NGS)は、多様な生物学的探究に取り組む研究者によってますます活用されるようになっている。最新のシーケンシングの驚くべきスケールと効率を活用し、ゲノム解析からタンパク質-核酸相互作用の複雑なダイナミクスに至るまで、様々な分野で大きな進歩が見られる。NGSデータが豊富な生物学的情報を持っていることを認識し、国際塩基配列データベース共同体(INSDC)は、公開塩基配列データと関連するメタデータを収集・普及するために約40年前に設立された。National Genomics Data Center (NGDC)もまた、膨大な量の生配列データへのオープンアクセスを提供してきた。これらのデータベースはNGSデータの再解析能力を大きく向上させた。近年、大規模言語モデルの台頭の中で、生物学的な課題に対処するためのモデルをトレーニングするためのインプットとして、生物学的な配列とデータが登場している。しかし、このような公開シーケンスデータにプログラムでアクセスする方法は依然として限られている。このギャップに対処するため、本著者らは、コマンドラインインターフェースを介してメタデータとNGSデータを迅速かつ簡単に検索できる統合ツール、iSeqを開発した。iSeqは現在、複数のデータベース(GSA、SRA、ENA、DDBJ、GEO)からの同時検索をサポートする唯一のツールである。さらに、iSeqは入力として幅広いアクセッション形式をサポートし、並列ダウンロード、マルチスレッド処理、FASTQファイルマージ機能を備えている。iSeqはBioconda(https://anaconda.org/bioconda/iseq)とGitHub(https://github.com/BioOmics/iSeq)から自由に利用できる。
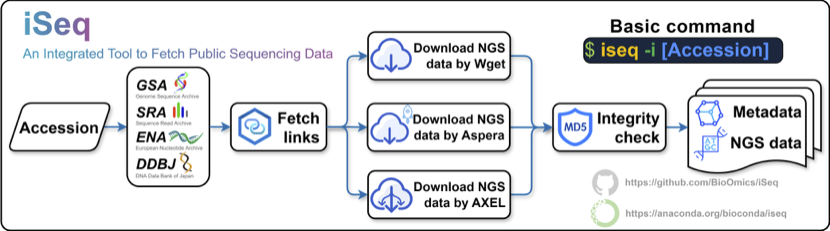
workflow. レポジトリより転載
インストール
ubuntu22で環境を作ってテストした。
#conda(link)
mamba create -n iseq -c conda-forge -c bioconda iseq
conda activate iseq
>iseq
$ iseq -h
Description:
Download sequencing data and matadata for each Run from GSA/SRA/ENA/DDBJ databases.
Usage:
iseq -i accession [options]
Input:
Accepted accession formats:
1. Projects: PRJEB, PRJNA, PRJDB, PRJC, GSE
2. Studies: ERP, DRP, SRP, CRA
3. BioSamples: SAMD, SAME, SAMN, SAMC
4. Samples: ERS, DRS, SRS, GSM
5. Experiments: ERX, DRX, SRX, CRX
6. Runs: ERR, DRR, SRR, CRR
Options:
Required parameter:
-i, --input TEXT accession (Project, Study, Sample, Experiment, or Run)
Optional parameters:
-m, --metadata Skip the sequencing data downloads and only fetch the metadata for the accession.
-g, --gzip Download FASTQ files in gzip format directly (*.fastq.gz).
note: if *.fastq.gz files are not available, SRA files will be downloaded and converted to *.fastq.gz files.
-q, --fastq Convert SRA files to FASTQ format.
-t, --threads INT The number of threads to use for converting SRA to FASTQ files or compressing FASTQ files (default: 8).
-e, --merge Merge multiple fastq files into one fastq file for each Experiment, the accession can't be the Run ID.
-d, --database [ena|sra] The database to download SRA files from (default: auto-detect),
note: some SRA files may not be available in the ENA database, even if you specify "ena".
-p, --parallel INT Download sequencing data in parallel, the number of connections needs to be specified, such as -p 10.
note: breakpoint continuation cannot be shared between different numbers of connections.
-a, --aspera Use Aspera to download sequencing data, only support GSA/ENA database.
-h, --help Show the help information.
-v, --version Show the script version.
See example:
https://github.com/BioOmics/iSeq/blob/main/docs/Examples.md
More information:
https://github.com/BioOmics/iSeq
実行方法
SRAの全サンプルを取得する。-gをつけるとfastqはgzip圧縮して".fastq.gz"として保存される。ペアエンドのシークエンシングデータはデフォルトで2つに分けて保存される。wgetが遅い場合、"-p"パラメータを使用してマルチスレッドでダウンロードできる。
iseq -i PRJNA211801 -g -p 6
- -g Download FASTQ files in gzip format directly (*.fastq.gz).
note: if *.fastq.gz files are not available, SRA files will be downloaded and converted to *.fastq.gz files. - -p Download sequencing data in parallel, the number of connections needs to be specified, such as -p 10.
実行時は進捗も表示される。
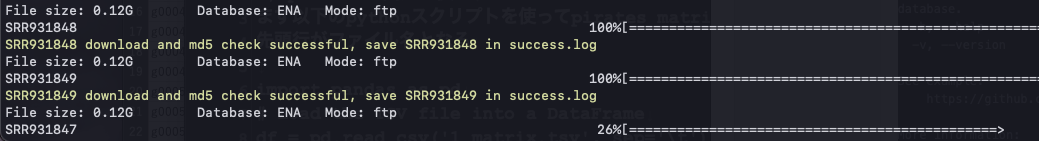
出力

Run IDを指定する。単独のシークエンシングデータのダウンロードになるので、"-e"オプションと互換性がない。
iseq -i DRR508037 -g
さまざまなオプションが用意されています。順番に見ていきます。
メタデータの取得 - メタデータ(上の画像のTSVファイル)のみダウンロードするするには”-m”をつける。
iseq -i PRJNA211801 -m
- -m Skip the sequencing data downloads and only fetch the metadata for the accession.
(レポジトリより: "-m"を使用するかどうかに関係なくそのアクセッションのサンプル情報は取得される。)
マージして保存 - fastqをマージして保存する。これは1つのExperimentに複数のRunが含まれる時を想定している(ペアエンドは分けて保存される)。
iseq -i SRX003906 -e -g
- -e Merge multiple fastq files into one fastq file for each Experiment, the accession can't be the Run ID.
(レポジトリより: アクセッションがRun IDの場合、-eパラメーターは使用できない。またBAMファイルやtar.gzファイルなどのファイルのマージはサポートされていない。)
データベース指定 - デフォルトでは、iSeqは利用可能なデータベースを自動的に検出するが、SRAファイルによってはENAデータベースからのダウンロードが遅い場合がある。このような場合、"-d sra"を指定してSRAデータベースから強制的にダウンロードする。
iseq -i SRR1178105 -d sra
- -d [ena | sra] The database to download SRA files from (default: auto-detect),
note: some SRA files may not be available in the ENA database, even if you specify "ena".
Asperaの使用 - IBMのAspera Connectを使うには-aをつける。
iseq -i PRJNA211801 -a -g
- -a Use Aspera to download sequencing data, only support GSA/ENA database.
(レポジトリより: Asperaを使ったダウンロードは現在GSAおよびENAデータベースでのみサポートされている。NCBI SRAデータベースは主にGoogle CloudとAWS Cloudテクノロジーを採用しているため、ダウンロードにAsperaを利用することができない。また、Asperaはキーファイルを必要とするため、iSeqは自動的にconda環境または~/.asperaディレクトリからキーファイルを検索する。キーファイルが見つからない場合、ダウンロードはできない。)

出力例
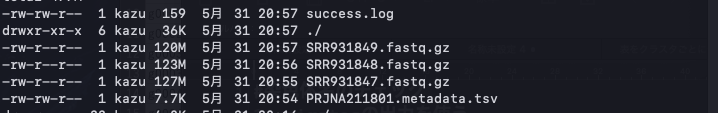
その他
- ダウンロードに対応するデータベースの一覧と類似ツールとの違いはレポジトリの表を参照
引用
iSeq: An integrated tool to fetch public sequencing data
Haoyu Chao, Zhuojin Li, Dijun Chen, Ming Chen
bioRxiv, Posted May 22, 2024.
関連
・SRAなどのシーケンシングデータを一括ダウンロードする grabseqs
参考