メタゲノムアセンブリゲノム(MAG)用のシーケンス比較ツールは、大量のデータや低品質のデータに対処するのが困難である。本著者らは、疎な近似アラインメントを用いて平均ヌクレオチド同一性(ANI)を決定する手法であるskani(https://github.com/bluenote-1577/skani)を提案する。skaniは、断片化され不完全なMAGsにおいて、FastANIよりも精度と速度(20倍以上高速)で優れている。skaniは、65,000を超える原核生物ゲノムに対して数秒で照合を実行し、6GBのメモリで動作する。skaniは、大規模でノイズの多いメタゲノムデータセットに対して、より高解像度の洞察を可能にする。
skani v0.3.0 is released. https://t.co/dEkIzxIbDr
— Jim Shaw (@jim_elevator) August 13, 2025
* 30-40% potential reduction in memory
* Breaking changes to indexing and searching databases
Calculate ANI for contigs, genomes. Search vs > 140k genomes: pre-indexed GTDB-R226 available for download.
basic usage guide
skani basic usage guide · bluenote-1577/skani Wiki · GitHub
インストール
ビルド依存
- rust programming language and associated tools such as cargo are required and assumed to be in PATH.
- A c compiler (e.g. GCC)
- make
git clone https://github.com/bluenote-1577/skani
cd skani
# If default rust install directory is ~/.cargo
cargo install --path . --root ~/.cargo
#conda
mamba install -c bioconda skani
#binary
wget https://github.com/bluenote-1577/skani/releases/download/latest/skani
chmod +x skani
./skani -h
> skani
skani 0.3.0
fast, robust ANI calculation and database searching for metagenomic contigs and assemblies.
Quick ANI calculation:
skani dist genome1.fa genome2.fa
Memory-efficient database search:
skani sketch genomes/* -o database; skani search -d database query1.fa query2.fa ...
All-to-all comparison:
skani triangle genomes/*
USAGE:
skani <SUBCOMMAND>
OPTIONS:
-h, --help Print help information
-V, --version Print version information
SUBCOMMANDS:
dist Compute ANI for queries against references fasta files or pre-computed sketch
files. Usage: skani dist query.fa ref1.fa ref2.fa ... or use -q/--ql and -r/--rl
options
search Search queries against a large pre-sketched database of reference genomes in a
memory efficient manner. Usage: skani search -d sketch_folder query1.fa
query2.fa ...
sketch Sketch (index) genomes. Usage: skani sketch genome1.fa genome2.fa ... -o
new_sketch_folder
triangle Compute a lower triangular ANI/AF matrix. Usage: skani triangle genome1.fa
genome2.fa genome3.fa ...
> skani dist -h
skani-dist
Compute ANI for queries against references fasta files or pre-computed sketch files. Usage: skani
dist query.fa ref1.fa ref2.fa ... or use -q/--ql and -r/--rl options
USAGE:
skani dist [OPTIONS] <QUERY|-q <QUERIES>...|--ql <QUERY_LIST>> [--] [REFERENCE]...
OPTIONS:
-h, --help Print help information
-t <THREADS> Number of threads [default: 3]
INPUTS:
-q <QUERIES>... Query fasta(s) or sketch(es)
--qi Use individual sequences for the QUERY in a multi-line fasta
--ql <QUERY_LIST> File with each line containing one fasta/sketch file
-r <REFERENCES>... Reference fasta(s) or sketch(es)
--ri Use individual sequences for the REFERENCE in a multi-line fasta
--rl <REFERENCE_LIST> File with each line containing one fasta/sketch file
<QUERY> Query fasta or sketch
<REFERENCE>... Reference fasta(s) or sketch(es)
OUTPUT:
-o <OUTPUT> Output file name; rewrites file by default [default: output
to stdout]
--min-af <MIN_AF> Only output ANI values where one genome has aligned fraction
> than this value. [default: 15]
--both-min-af <BOTH_MIN_AF> Only output ANI values where both genomes have aligned
fraction > than this value. [default: disabled]
--ci Output [5%,95%] ANI confidence intervals using percentile
bootstrap on the putative ANI distribution
--detailed Print additional info including contig N50s and more
-n <N> Max number of results to show for each query. [default:
unlimited]
--short-header Only display the first part of contig names (before first
whitespace)
PRESETS:
--fast Faster skani mode; 2x faster and less memory. Less accurate AF and less
accurate ANI for distant genomes, but works ok for high N50 and > 95%
ANI. Alias for -c 200
--medium Medium skani mode; 2x slower and more memory. More accurate AF and more
accurate ANI for moderately fragmented assemblies (< 10kb N50). Alias for
-c 70
--slow Slower skani mode; 4x slower and more memory. Gives much more accurate AF
for distant genomes. More accurate ANI for VERY fragmented assemblies (<
3kb N50), but less accurate ANI otherwise. Alias for -c 30
--small-genomes Mode for small genomes such as viruses or plasmids (< 20 kb). Can be much
faster for large data, but is slower/less accurate on bacterial-sized
genomes. Alias for: -c 30 -m 200 --faster-small
ALGORITHM PARAMETERS:
-c <C> Compression factor (k-mer subsampling rate). [default: 125]
--faster-small Filter genomes with < 20 marker k-mers more aggressively. Much faster
for many small genomes but may miss some comparisons
-m <MARKER_C> Marker k-mer compression factor. Markers are used for filtering.
Consider decreasing to ~200-300 if working with small genomes (e.g.
plasmids or viruses). [default: 1000]
--median Estimate median identity instead of average (mean) identity
--no-learned-ani Disable regression model for ANI prediction. [default: learned ANI used
for c >= 70 and >= 150,000 bases aligned and not on individual contigs]
--no-marker-index Do not use hash-table inverted index for faster ANI filtering.
[default: load index if > 100 query files or using the --qi option]
--robust Estimate mean after trimming off 10%/90% quantiles
-s <S> Screen out pairs with *approximately* < % identity using k-mer
sketching. [default: 80]
MISC:
--trace Trace level verbosity
-v, --debug Debug level verbosity
> skani search -h
skani-search
Search queries against a large pre-sketched database of reference genomes in a memory efficient
manner. Usage: skani search -d sketch_folder query1.fa query2.fa ...
USAGE:
skani search [OPTIONS] -d <DATABASE> <QUERY|-q <QUERIES>...|--ql <QUERY_LIST>> [--]
OPTIONS:
-h, --help Print help information
-t <THREADS> Number of threads [default: 3]
INPUTS:
-d <DATABASE> Output folder from `skani sketch`
-q <QUERIES>... Query fasta(s) or sketch(es)
--qi Use individual sequences for the QUERY in a multi-line fasta
--ql <QUERY_LIST> File with each line containing one fasta/sketch file
<QUERY>... Query fasta(s) or sketch(es)
OUTPUT:
-o <OUTPUT> Output file name; rewrites file by default [default: output
to stdout]
--both-min-af <BOTH_MIN_AF> Only output ANI values where both genomes have aligned
fraction > than this value. [default: disabled]
--ci Output [5%,95%] ANI confidence intervals using percentile
bootstrap on the putative ANI distribution
--detailed Print additional info including contig N50s and more
--min-af <MIN_AF> Only output ANI values where one genome has aligned fraction
> than this value. [default: 15]
-n <N> Max number of results to show for each query. [default:
unlimited]
--short-header Only display the first part of contig names (before first
whitespace)
ALGORITHM PARAMETERS:
--keep-refs Keep reference sketches in memory if the sketch passes the marker
filter. Takes more memory but is much faster when querying many similar
sequences
--median Estimate median identity instead of average (mean) identity
--no-learned-ani Disable regression model for ANI prediction. [default: learned ANI used
for c >= 70 and >= 150,000 bases aligned and not on individual contigs]
--no-marker-index Do not use hash-table inverted index for faster ANI filtering.
[default: load index if > 100 query files or using the --qi option]
--robust Estimate mean after trimming off 10%/90% quantiles
-s <S> Screen out pairs with *approximately* < % identity using k-mer
sketching. [default: 80]
MISC:
--trace Trace level verbosity
-v, --debug Debug level verbosity
> skani triangle -h
skani-triangle
Compute a lower triangular ANI/AF matrix. Usage: skani triangle genome1.fa genome2.fa genome3.fa ...
USAGE:
skani triangle [OPTIONS] <-l <FASTA_LIST>|FASTA_FILES>
OPTIONS:
-h, --help Print help information
-t <THREADS> Number of threads [default: 3]
INPUTS:
-i Use individual sequences instead the entire file for multi-fastas
-l <FASTA_LIST> File with each line containing one fasta/sketch file
<FASTA_FILES>... Fasta(s) or sketch(es)
OUTPUT:
-o <OUTPUT> Output file name; rewrites file by default [default: output
to stdout]
--both-min-af <BOTH_MIN_AF> Only output ANI values where both genomes have aligned
fraction > than this value. [default: disabled]
--ci Output [5%,95%] ANI confidence intervals using percentile
bootstrap on the putative ANI distribution. Only works with
--sparse or -E
--detailed Print additional info including contig N50s and more
--diagonal Output the diagonal of the ANI matrix (i.e. self-self
comparisons) for both dense and sparse matrices
--distance Output 100 - ANI instead of ANI, creating a distance instead
of a similarity matrix. No effect if using --sparse or -E
-E, --sparse Output comparisons in a row-by-row form (i.e. sparse matrix)
in the same form as `skani dist`. Only pairs with aligned
fraction > --min-af are output
--full-matrix Output full matrix instead of lower-triangular matrix
--min-af <MIN_AF> Only output ANI values where one genome has aligned fraction
> than this value. [default: 15]
--short-header Only display the first part of contig names (before first
whitespace)
PRESETS:
--fast Faster skani mode; 2x faster and less memory. Less accurate AF and less
accurate ANI for distant genomes, but works ok for high N50 and > 95%
ANI. Alias for -c 200
--medium Medium skani mode; 2x slower and more memory. More accurate AF and more
accurate ANI for moderately fragmented assemblies (< 10kb N50). Alias for
-c 70
--slow Slower skani mode; 4x slower and more memory. Gives much more accurate AF
for distant genomes. More accurate ANI for VERY fragmented assemblies (<
3kb N50), but less accurate ANI otherwise. Alias for -c 30
--small-genomes Mode for small genomes such as viruses or plasmids (< 20 kb). Can be much
faster for large data, but is slower/less accurate on bacterial-sized
genomes. Alias for: -c 30 -m 200 --faster-small
ALGORITHM PARAMETERS:
-c <C> Compression factor (k-mer subsampling rate). [default: 125]
--faster-small Filter genomes with < 20 marker k-mers more aggressively. Much faster
for many small genomes but may miss some comparisons
-m <MARKER_C> Marker k-mer compression factor. Markers are used for filtering.
Consider decreasing to ~200-300 if working with small genomes (e.g.
plasmids or viruses). [default: 1000]
--median Estimate median identity instead of average (mean) identity
--no-learned-ani Disable regression model for ANI prediction. [default: learned ANI used
for c >= 70 and >= 150,000 bases aligned and not on individual contigs]
--robust Estimate mean after trimming off 10%/90% quantiles
-s <S> Screen out pairs with *approximately* < % identity using k-mer
sketching. [default: 80]
MISC:
--trace Trace level verbosity
-v, --debug Debug level verbosity
> skani sketch -h
skani-sketch
Sketch (index) genomes. Usage: skani sketch genome1.fa genome2.fa ... -o new_sketch_folder
USAGE:
skani sketch [OPTIONS] -o <OUTPUT> <FASTA_FILES|-l <FASTA_LIST>>
OPTIONS:
-h, --help Print help information
-t <THREADS> Number of threads [default: 3]
INPUT/OUTPUT:
-o <OUTPUT> Output folder where sketch files are placed
-i Use individual sequences instead the entire file for multi-fastas
-l <FASTA_LIST> File with each line containing one fasta/sketch file
--separate-sketches Create separate .sketch files instead of consolidated database
format. DOES NOT WORK WITH -i
<FASTA_FILES>... fastas to sketch
PRESETS:
--fast Faster skani mode; 2x faster and less memory. Less accurate AF and less accurate
ANI for distant genomes, but works ok for high N50 and > 95% ANI. Alias for -c
200
--medium Medium skani mode; 2x slower and more memory. More accurate AF and more accurate
ANI for moderately fragmented assemblies (< 10kb N50). Alias for -c 70
--slow Slower skani mode; 4x slower and more memory. Gives much more accurate AF for
distant genomes. More accurate ANI for VERY fragmented assemblies (< 3kb N50),
but less accurate ANI otherwise. Alias for -c 30
SKETCH PARAMETERS:
-c <C> Compression factor (k-mer subsampling rate). [default: 125]
-m <MARKER_C> Marker k-mer compression factor. Markers are used for filtering. Consider
decreasing to ~200-300 if working with small genomes (e.g. plasmids or
viruses). [default: 1000]
MISC:
--trace Trace level verbosity
-v, --debug Debug level verbosity
(バージョンに注意する。古いバージョンで作成されたスケッチファイルは認識しない。)
実行方法
2つのゲノムを比較するには2つのfastaファイルを順番に指定する(順番は結果に影響しない)。
git clone https://github.com/bluenote-1577/skani.git
cd skani/test_files/
skani dist e.coli-EC590.fasta e.coli-K12.fasta
出力例

ANI 99.39, Alignment fraction 91.89
複数ゲノム間の比較、結果はresults.txtに書き出し。
skani dist -t 3 -q e.coli-W.fasta e.coli-K12.fasta -r e.coli-EC590.fasta e.coli-W.fasta -o results.txt
#ワイルドカードにも対応
skani dist -t 3 -q genome*fa -r e.coli-EC590.fasta -o results.txt
出力例

データベースを構築して使用。-dでDBのディレクトリを指定する。
skani sketch fasta/* -o database
# => database/ができる
#データベースと比較
skani search query1.fa query2.fa -d database
出力例

フォルダ内全ゲノムペア間のANI行列の作成。-Eをつけるとスパース形式(行ごとにペアを表示)となる。--min-afと組み合わせて意味のあるANI値だけを書き出す。
skani triangle fasta/* > skani_ani_matrix.txt
- -E Output comparisons in a row-by-row form (i.e. sparse matrix) in the same form as `skani dist`. Only pairs with aligned fraction > --min-af are output
- --full-matrix Output full matrix instead of lower-triangular matrix
- --min-af <MIN_AF> Only output ANI values where one genome has aligned fraction
出力例

マトリクスファイルからクラスタリングされたヒートポンプに可視化するスクリプトが用意されている(python3, seaborn, scipy/numpy, and matplotlibが必要)。
python scripts/clustermap_triangle.py skani_ani_matrix.txt
GTDB R226のPre-sketched databasesをダウンロードしてsketched databaseとして使用する事ができる。
wget http://faust.compbio.cs.cmu.edu/skani-files/skani_gtdb_r226-v0.3.tar.gz
tar -zxvf skani_gtdb_r226-v0.3.tar.gz
skani search my_genome.fa -d skani_gtdb_r226-v0.3 -o results.tsv
tar.gzファイルサイズは37GB。解凍後は57GBディスクスペースを占有する。ゲノム1個だけで問い合わせたところ、22秒かかった(E5 2680 v4)。
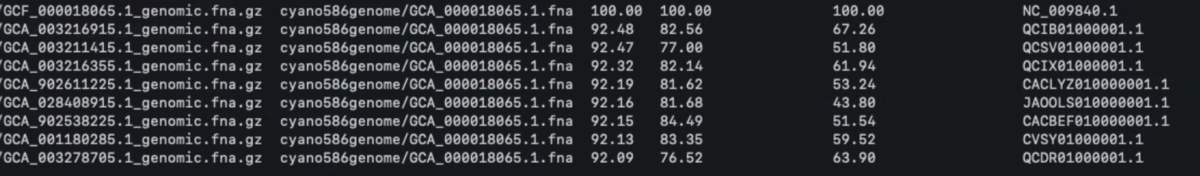
結果はペアワイズyANI値が高い順に並んでいる。GTDB rep. DBには各種の代表ゲノムは1個ずつしか含まれていないので、トップヒットのみ返すoptionがあってもいいかもしれない(クエリと同種を調べる目的ならこれで達成できる)。
その他
- skaniは、塩基レベルのアライメントを行わない近似マッピング法を用いてDNA配列(コンティグ/MAG/ゲノム)およびANI > 約80%の平均ヌクレオチド同一性(ANI)と整列の割合(AF)を計算する。
- 純粋なスケッチング手法(例:Mash)は不完全なMAGのANIを過小評価することがあるが、skaniは不完全かつ中程度の品質のメタゲノムアセンブリゲノム(MAG)に対しても正確。
- インデックス作成/スケッチングはMashより約3倍高速で、クエリ処理はFastANIより約25倍高速(ただしMashよりは遅い)
- 効率的なデータベース検索。事前処理された65,000以上の原核生物ゲノムのデータベースに対して、単一プロセッサと約6GBのRAMで数秒でクエリ検索が可能。ゲノム配列からデータベースを構築するのには数分から1時間程度かかる。
- v0.3.0では古い.sketchファイルは使用できなくなっているので注意する。
- v0.3.0ではデフォルトで個別の.sketchファイルではなく単一のデータベースが作成される。以前の動作は--separate-sketchesオプションで再現可能。
- -cで精度と感度を調整できる。デフォルトは-c 125となっている。
- --slow(-c 30):小さく断片化した / ANIが低いゲノム向け。--medium(-c 70):中程度に断片していたり距離のあるゲノム向け。高品質ゲノムではANIを少し過小評価する可能性がある。--fast(-c 200):高速・小メモリ使用量。10kb以上のN50やANI > 95%の場合に適し、AF が不要であれば選択可能。
- ウィルスと小さなプラスミド向けに--small-genomes オプションが用意されている。これは-c 30 -m 200 --faster-small を一括して設定する。3kb以上のウイルスを扱う場合は -m 150などを考える。
-
-s オプションでANI値の閾値をセットする。デフォルトは 80%で、より厳密に近縁ゲノム間のみ比較したい場合、-s を高く設定することができる。
- デフォルト設定では ANI82%以上までが信頼性が高い結果が得られる(AF > 15% が条件)。
引用
Fast and robust metagenomic sequence comparison through sparse chaining with skani
Jim Shaw & Yun William Yu
Nature Methods volume 20, pages1661–1665 (2023)
関連