2021 8/5追記、9/6 追記、10/8 contigコマンド修正
2022/05/09 help修正、06/03 コマンド
2023/08/10 追記
2024/04/12 構成を整頓, 7/20 thread option追記
2025/06/01追記、6/6 誤次修正
Githubより
CoverMは、メタゲノミクスアプリケーションに特化した、設定可能で使いやすく、高速なDNAリードカバレッジおよび相対的な存在比の計算ツールである。ゲノム/MAGのカバレッジをまたは個々のコンティグのカバレッジを計算する。入力は、リファレンスごとにソートされたBAMファイル、または様々なフォーマットの生のリードとリファレンスゲノムのいずれかである。
HP
https://wwood.github.io/CoverM/coverm-genome.html#name
2025/04/10
Our newest tool, CoverM, a unified software package which calculates several coverage statistics for contigs and genomes in an ergonomic and flexible manner has been published in Bioinformatics.
— Centre for Microbiome Research (@CMR_QUT) April 10, 2025
You can find the full article at the link below!https://t.co/q3ppYI8xTu
2024/01/25
CoverM 0.7.0, metagenome coverage calculator, is out. Citation, strobealign for faster mapping, skani for faster genome derep, bwa-mem2 and checkm2 support. https://t.co/KCIeBh9IOU
— Ben WoOdcroft (@wwood) 2024年1月24日
Too many ppl to thank in short tweet..
インストール
ubuntu18.04LTSでテストした(linuxマシン使用)。
依存
- samtools v1.9
- tee, which is installed by default on most Linux operating systems.
- man, which is installed by default on most Linux operating systems.
some mapping software:
- minimap2 v2.17-r941 (2.18 will not work)
- bwa-mem2 v2.0
For dereplication:
- Dashing v0.4.0
- FastANI v1.3
#bioconda (link)
mamba install -c bioconda -y coverm
#cargoにも対応
cargo install coverm
> coverm
$ coverm
Mapping coverage analysis for metagenomics
Usage: coverm <subcommand> ...
Main subcommands:
contig Calculate coverage of contigs
genome Calculate coverage of genomes
Less used utility subcommands:
make Generate BAM files through alignment
filter Remove (or only keep) alignments with insufficient identity
cluster Dereplicate and cluster genomes
shell-completion
Generate shell completion scripts
Other options:
-V, --version Print version information
Ben J. Woodcroft <benjwoodcroft near gmail.com>
> coverm contig
$ coverm contig
error: The following required arguments were not provided:
--coupled <coupled>...
--interleaved <interleaved>...
-1 <read1>...
-2 <read2>...
--reference <reference>...
--single <single>...
USAGE:
coverm contig --contig-end-exclusion <contig-end-exclusion> --coupled <coupled>... --interleaved <interleaved>... --mapper <mapper> --methods <methods>... --min-covered-fraction <min-covered-fraction> --output-format <output-format> -1 <read1>... -2 <read2>... --reference <reference>... --single <single>... --threads <threads> --trim-max <trim-max> --trim-min <trim-min>
For more information try --help
(base) kazu@kazu:/media/kazu/optane/TY1$ coverm contig -h
coverm contig
Calculate coverage of individual contigs
Example: Calculate mean coverage from reads and assembly:
coverm contig --coupled read1.fastq.gz read2.fastq.gz --reference assembly.fna
Example: Calculate MetaBAT adjusted coverage from a sorted BAM file, saving
the unfiltered BAM files in the saved_bam_files folder:
coverm contig --method metabat --bam-files my.bam
--bam-file-cache-directory saved_bam_files
See coverm contig --full-help for further options and further detail.
> coverm genome -h
$ coverm genome -h
coverm genome
Calculate coverage of individual genomes
Example: Map paired reads to 2 genomes, and output relative abundances
to output.tsv:
coverm genome --coupled read1.fastq.gz read2.fastq.gz
--genome-fasta-files genome1.fna genome2.fna -o output.tsv
Example: Calculate coverage of genomes defined as .fna files in
genomes_directory/ from a sorted BAM file:
coverm genome --bam-files my.bam --genome-fasta-directory genomes_directory/
Example: Dereplicate genomes at 99% ANI before mapping unpaired reads:
coverm genome --genome-fasta-directory genomes/ --dereplicate
--single single_reads.fq.gz
See coverm genome --full-help for further options and further detail.
> coverm bam -h
$ coverm bam -h
error: Found argument 'bam' which wasn't expected, or isn't valid in this context
USAGE:
coverm [FLAGS] [SUBCOMMAND]
For more information try --help
> coverm filter -h
coverm filter
Filter BAM file alignments
Example: Filter a BAM file by removing alignments shorter than 50bp:
coverm filter --bam-files input.bam --output-bam filtered.bam
--min-read-aligned-length 50
Example: Filter inverse: Keep alignments that have <95% alignment identity
and those which do map at all. Note that the output BAM file will likely
records that are still mapped, but align with < 95% identity. Use 16
threads for output compression:
coverm filter -b input.bam -o inverse_filtered.bam --inverse
--min-read-percent-identity 95 --threads 16
See coverm filter --full-help for further options and further detail.
> coverm cluster -h
$ coverm cluster -h
coverm cluster
Cluster (dereplicate) genomes
Example: Dereplicate at 99% (after pre-clustering at 95%) a directory of .fna
FASTA files and create a new directory of symlinked FASTA files of
epresentatives:
coverm cluster --genome-fasta-directory input_genomes/
--output-representative-fasta-directory output_directory/
Example: Dereplicate a set of genomes with paths specified in genomes.txt at
95% ANI, after a preclustering at 90% using the MinHash finch method, and
output the cluster definition to clusters.tsv:
coverm cluster --ani 95 --precluster-ani 90 --precluster-method finch
--genome-fasta-list genomes.txt
--output-cluster-definition clusters.tsv
See coverm cluster --full-help for further options and further detail.
--full-helpをつけてコマンドを実行するとオプションの詳細が確認できる。
実行方法
coverm genome ゲノム単位のカバレッジ(ゲノムモード)の計算
ゲノムモードの計算は、コンティグ単位のカバレッジ(コンティグモード)の計算と似ているが、報告の単位がコンティグ単位ではなくゲノム単位である点が異なる。カバレッジに基づいて塩基の位置を除外する計算方法では、すべてのコンティグからのすべての位置が一緒に考慮される。例えば、2,000,000bpのコンティグがあり、そのコンティグの全ての位置のカバレッジが1Xである場合、2000bpの全ての位置がカバレッジでソートされた位置の上位5%に入るため、trimmed_meanは0となる。
A、マッピング済みのbamを指定
bin配列をcatで固めたリファレンスにマッピングしてbamファイルを作る(*1)。coverMを実行する時には、bamファイルとcatで固める前の各ゲノム配列のfastaを保存したディレクトリを指定する。fastaファイルの拡張子が.fnaなら”-x fna”とする。
#relative_abundance
coverm genome --bam-files input.bam --genome-fasta-directory bin_dir -x fna -m relative_abundance > out.tsv
#mean
coverm genome --bam-files input.bam --genome-fasta-directory bin_dir -x fna -m mean > out.tsv
#TPM
coverm genome --bam-files input.bam --genome-fasta-directory bin_dir -x fna -m tpm > out.tsv
#read count
coverm genome --bam-files input.bam --genome-fasta-directory bin_dir -x fna -m count --min-covered-fraction > out.tsv
-
-f, --genome-fasta-files Path(s) to FASTA files of each genome
-
-d, --genome-fasta-directory Directory containing FASTA files of each genome.
-
-x, --genome-fasta-extension File extension of genomes in the directory specified with -d/--genome-fasta-directory. [default: fna]
- --single-genome All contigs are from the same genome. Requires --reference. [default: not set]
- -r, --reference FASTA file of contigs e.g. concatenated genomes or metagenome assembly, or minimap2 index (with --minimap2-reference-is-index), or BWA index stem (with -p bwa-mem). If multiple references FASTA files are provided and
--sharded is specified, then reads will be mapped to references separately as sharded BAMs. NOTE: If genomic FASTA files are specified elsewhere (e.g. with --genome-fasta-files or --genome-fasta-directory), then --reference
is not needed as a reference FASTA file can be derived by concatenating input genomes. However, while not necessary, --reference can be specified if an alternate reference sequence set is desired. - -m, --methods Method(s) for calculating coverage [default: relative_abundance]. A more thorough description of the different methods is available at https://github.com/wwood/CoverM#calculation-methods
B、fastqを指定
マッピングにはminimap2が使用される。
#1組のペアエンド
coverm genome -1_R1.fq.gz -2 in_R2.fq.gz --genome-fasta-directory genome_dir -x fna -o out.tsv -t 20
#複数のペアエンドfastq
coverm genome --coupled sample1_R1.fq.gz sample1_R2.fq.gz sample2_R1.fq.gz sample2_R2.fq.gz sample3_R1.fq.gz sample3_R2.fq.gz --genome-fasta-directory genome_dir -x fna -o out.tsv -t 20
-
-1 Forward FASTA/Q file(s) for mapping. These may be gzipped or not.
-
-2 Reverse FASTA/Q file(s) for mapping. These may be gzipped or not.
-
-c, --coupled One or more pairs of forward and reverse possibly gzipped FASTA/Q files for mapping in order <sample1_R1.fq.gz> <sample1_R2.fq.gz> <sample2_R1.fq.gz> <sample2_R2.fq.gz> ..
- -t Number of threads for mapping, sorting and reading. [default: 1]
複数の定量方法がサポートされている。デフォルトではrelative_abundanceとなっている。"-m"オプションを使って変更できる。
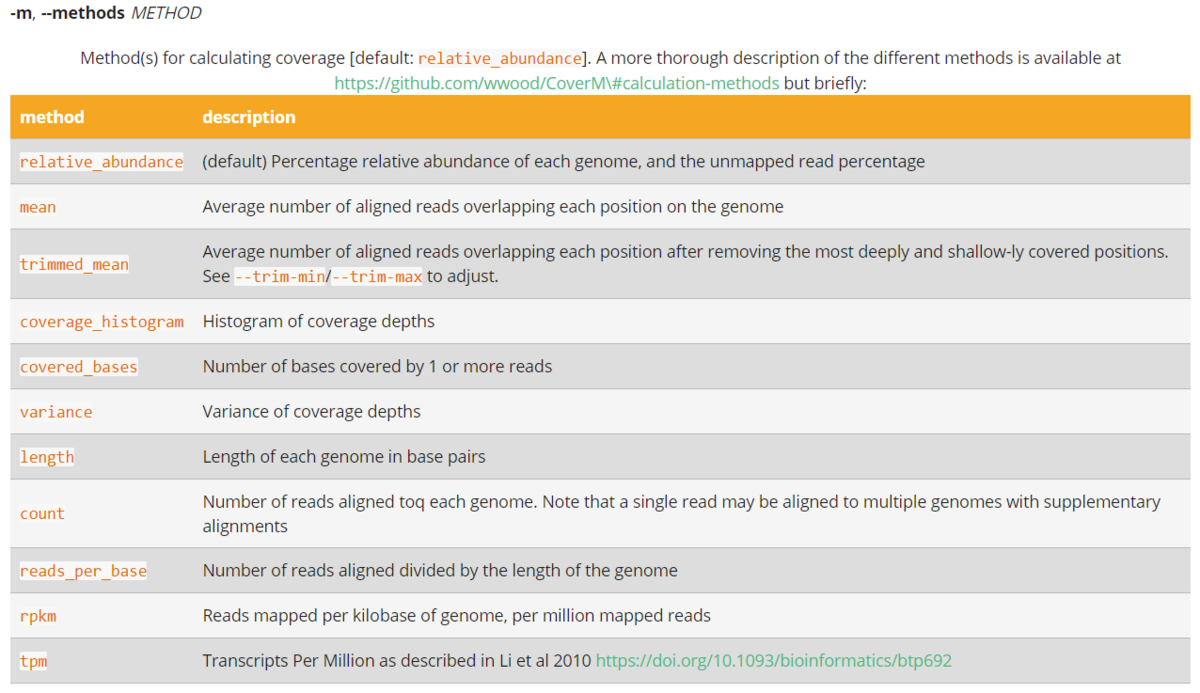
(HPより)
* ゲノムサイズの補正も行われます(2022/0906追記)
結果を整形し、階層的クラスタリングしてheatmapで視覚化する。
https://github.com/tillrobin/iMGMC/blob/master/tutorials/map-to-MAGs-HeatmapR.md
coverm filter identityが不十分なアラインメントを削除する。信頼性の低いマッピングをフィルタリングするために使える。
coverm filter -b input.bam -o filtered.bam --inverse --min-read-percent-identity 95 --threads 16
coverm contig コンティグ単位のカバレッジの計算
coverm contig --bam-files input.bam
coverm make アライメントによるBAMファイルの生成
coverm cluster ゲノムのdereplicationとクラスター化
その他
- coverm genomeではANI基準でdereplicationを行うオプションも存在する。詳細は"coverm genome --full-help"で確認できる。
- coerMのパラメータ例;https://www.nature.com/articles/s41467-021-22203-2
- 大量のfastqファイルをリストで指定(#149)することはできないので、1つずつランし、後で結合する。デフォルトのrelative_abundance正規化に影響はない。
- ジョブはシングルスレッドらしい。大量にfastqがあるときは並列ランする(ピークメモリは配列数で変わる。1000個のMAGだと20~30giga byesくらい)。
- *1 複数のbin.fastaをcatで結合したファイルをリファレンスとしてマッピングする時は、minimap2をあらかじめindexingしてから使うのが高速。ところで、多くのMAGを固めたリファレンス(>100)をminimap2でindexを作ってマッピングすると、実際の外環境を読んだfastqを使った時に稀に10~30倍速度が低下する現象が確認される(v.2.26使用)。再現性が不明だが、全くマッピングされないfastqではこの現象は起きず、稀にマッピングされるMAGリファレンスが含まれるfastqを使った時、時々起こるようである(遭遇頻度は5%以下)。マッピング速度が安定しているbwa-mem2がお勧め。ただしメモリ使用量がかなり高いので注意したい。bowtie2の”sensitive setting”と比べてもマッピング感度は高く、minimap2とbwa memはbowtie2の2-3倍のリードがアラインされることもある(差が出るのはリファレンスから距離があるリアルメタデータに限られる)。minimap2とbwa memを比べると、わずかにbwa memの方がアラインされるリードは多い傾向(大規模な比較ではないので注意)。
- 作業中、coverm genomeなどはシステムディスクに数十GBの作業領域を確保する(/tmp)。たくさんのサンプルを扱う時は、システムディスクが一杯にならないようにTMPDIR環境変数を空容量の多いディスクに変更する。例えば、
mkdir media/kazu/8TBHDD1/tmp
export TMPDIR=/media/kazu/8TBHDD1/tmp
のようにする。
引用
GitHub - wwood/CoverM: Read coverage calculator for metagenomics
2025/04/10 追記
CoverM: Read alignment statistics for metagenomics
Samuel T N Aroney, Rhys J P Newell, Jakob N Nissen, Antonio Pedro Camargo, Gene W Tyson, Ben J Woodcroft
Bioinformatics, Published: 07 April 2025
関連
オススメ