GSE(Gene Set Enrichment)解析は、ゲノムスケールの実験から生物学的な知見を引き出すために重要な役割を担っている。ORA (overrepresentation analysis)、FCS (functional class scoring)、PT (pathway topology) のアプローチは、GSE手法の3世代に渡って発展してきたものである。KOBASの旧バージョンでは、ORA法のみをベースにしたサービスを提供していた。今回、KOBASのバージョン3.0を発表し、KOBAS-i(KOBAS intelligent versionの略)と名づけた。これは、7つのFCSツールと2つのPTツールを1つのアンサンブルスコアに統合し、関連する生物パスウェイをインテリジェントに優先順位付けするもので、先に発表した機械学習ベースの新しい手法、CGPSを導入している。さらに、KOBASはエンリッチされた結果を選択し、理解するための下流の探索的可視化機能を拡張した。このツールは、cirFunMapの新しいビューを構築し、異なるエンリッチされた語彙とその相関関係をランドスケープで表示する。最後に、前バージョンのフレームワークに基づき、KOBASはサポートする生物種を1327種から5944種に増やした。また、より簡単にローカルで実行できるように、ソースコード版の補足として、インストール不要のビルド済みDockerイメージも提供している。KOBASはhttp://kobas.cbi.pku.edu.cn、ミラーサイトはhttp://bioinfo.org/kobasから自由にアクセスできる。
Downloadからスタンドアローン版をダウンロードできる。
http://kobas.cbi.pku.edu.cn/download/
http://kobas.cbi.pku.edu.cn/にアクセスする。

KOBASは、アノテーションモジュールとエンリッチメントモジュールから構成されている。アノテーションモジュールは、IDや配列を含む遺伝子リストを入力とし、パスウェイ、疾患、GO情報などの複数のデータベースを基に、各遺伝子のアノテーションを生成する。エンリッチメントモジュールは、入力された遺伝子リストや発現と統計的に有意に関連するパスウェイやGOタームについて報告する。
Annotationタブ
Annotationタブに移動する。アノテーションモジュールではID や配列を含む遺伝子/タンパク質リストを入力として受け付ける。

種を選択する。

Input typeを選択する。

遺伝子リストを入力する。入力について、パスウェイや疾患、Gene Ontologyの語彙などの複数のデータベースに基づいて、各遺伝子のアノテーションが作成される。ここではexampleを指定。ヒトの遺伝子シンボルとなってる。

ファイルをドラッグして指定することもできる。
出力(example)
pathway、disease、GOに分かれている。
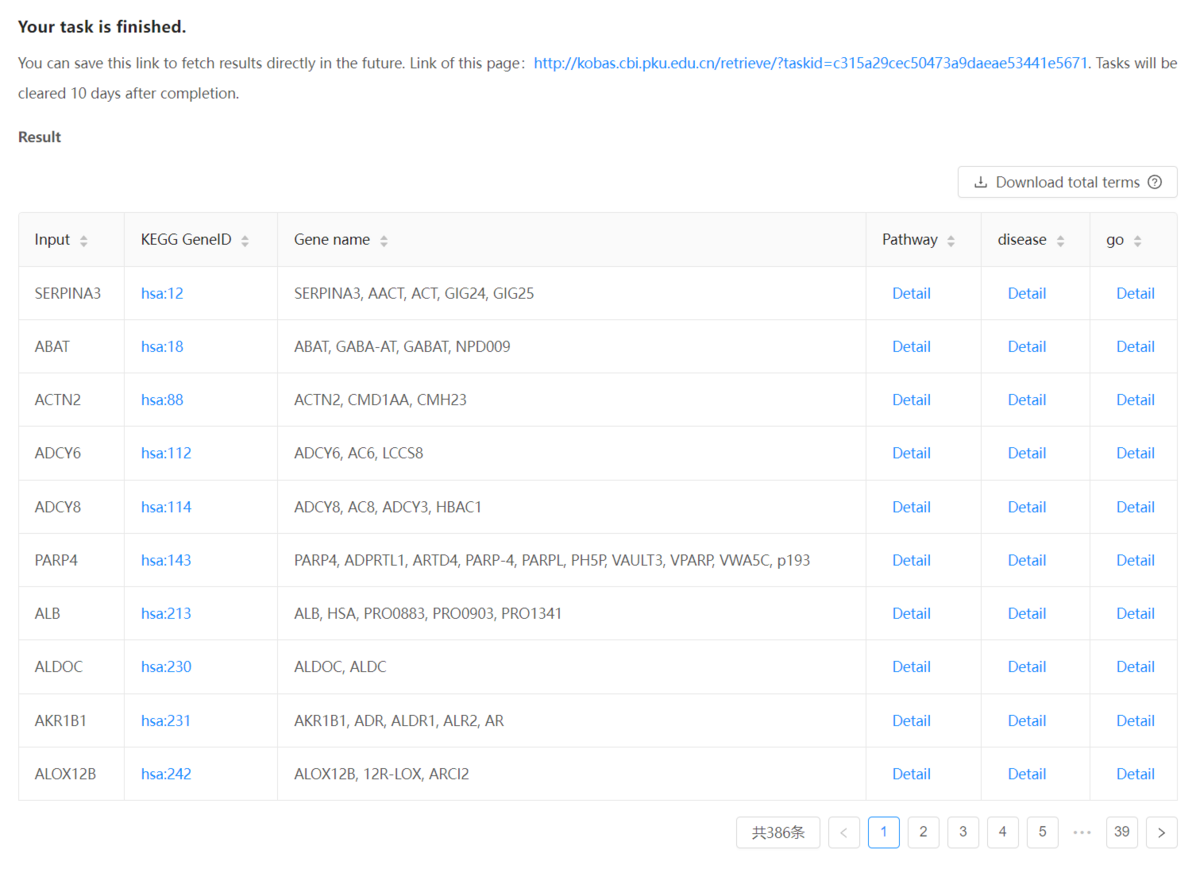
Detailををクリックすると詳細が表示される。

パスウェイはKEGG poathwayに、GOはAmiGO2にリンクしている。結果はテキスト形式でダウンロードできる。
Enrichmentタブ
入力形式の違いにより、2つのモジュールがある。

1,遺伝子リストエンリッチメント
KOBAS 2.0 では Identify と呼ばれている。アノテーションモジュールと同じ入力形式を受け付け、アノテーションモジュールの結果を入力として使用することができる。Hypergeometric test と Fisher's exact test を用いたORA (overrepresentation analysis) 法というシンプルでよく使われる遺伝子セット濃縮法がベースになっている。DAVIDをはじめ、様々なツールがこの方法を適用している。ただし、二項検定、カイ二乗検定、頻度リスト、Benjamini and Hochberg, Benjamini and Yekutieli, QVALUE などの3つのFDR補正法もサポートされている(マニュアルより)。
2,Exp-data Enrichment (エクスプデータ・エンリッチメント)
KOBAS 3.0 の新機能。遺伝子発現を入力とすることで、機能的な遺伝子集合の濃縮(GSE)解析に大きな変化をもたらす。ORA法では避けられなかった、特定の閾値を設定して発現量の多い、あるいは少ない差分遺伝子を選択するのではなく、特定の遺伝子セット内の全ての遺伝子の発現量を用いて機能スコアを算出する。
本モジュールでは、ORA法を含む9つの手法を統合した。Globaltest, GSEA, GSA PADOG, PLAGE, GAGE, SAFE, PT-based methods: GANPA, CEPA
さらに、Exp-data Enrichmentモジュールは、複数の手法でサポートされたエンリッチな遺伝子セットを検出するために、9つの遺伝子セットエクステンション(GSE)手法の結果を基に、アンサンブルスコア(R score)とエンリッチセットである確率を提供する(マニュアルより)。
ここでは1のGene-list Enrichmentを選んだ。
遺伝子リストとパスウェイデータベースを指定する。

ランする。
出力

Visualization
クリックすると数十秒で結果が表示される。
各ノードはエンリッチされたtermを表し、色は異なるクラスターを表す。ノードのサイズはエンリッチされたp値の6つのレベルで表している。

各行はエンリッチされた機能を表し、棒の長さは「入力遺伝子数」/「バックグランド遺伝子数」として計算されたエンリッチ比を表す。各クラスタにおいて、5つ以上のタームがある場合、最もエンリッチ比の高い上位5つが表示される。色は異なるクラスターを表す。

各バブルはエンリッチされた機能を表し、バブルの大きさはエンリッチされたp値の6つのレベルで表している。
引用
KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis
Dechao Bu, Haitao Luo, Peipei Huo, Zhihao Wang, Shan Zhang, Zihao He, Yang Wu, Lianhe Zhao, Jingjia Liu, Jincheng Guo, Shuangsang Fang, Wanchen Cao, Lan Yi, Yi Zha, Lei Kong
Nucleic Acids Res. 2021 Jul 2;49(W1):W317-W325
関連