2024/09/29 修正
パンゲノム解析は、細菌集団で起こる分子進化を探るための基本的な手法である。ここでは、パンゲノムデータを簡単に扱うことができるRフレームワークPagooを紹介する。Pagooはカプセル化されているため、複雑な分子情報や表現型情報をオブジェクト指向のアプローチで保存することができる。これにより、1つのプログラミング環境を用いてデータを遡ったり戻したりすることが容易になり、解析のどの段階でも(生データを含めて)1つのファイルに保存することができるため、共有可能で再現性のあるものとなる。Pagooは、Rエコシステムで利用可能な他の微生物ゲノム解析パッケージと連携して、クエリー、サブセット、比較、可視化、統計解析を実行するツールを提供する。実用例として、Pagooがスケーラブルであることを示すために1,000個の大腸菌ゲノムを使用した。また、病原体における進化のパターンと宿主適応のゲノムマーカーを同定するためにカンピロバクター・フェタスのゲノムのグローバルデータセットを使用した。
reference
https://iferres.github.io/pagoo/reference/index.html
Get started
https://iferres.github.io/pagoo/articles/Methods_Plots.html
インストール
ubuntu22でテストした(R v.4.2.2使用)。
install.packages("pagoo")
#install the latest dev from repository
if (!require("devtools")) install.packages("devtools")
devtools::install_github('iferres/pagoo')
実行方法
pagooを使うことで、パンゲノムの基本的な基本的な統計解析と可視化をすぐに行うことができます。チュートリアルに沿って手順を簡単に確認します。
1,デモデータの読み込み
library(pagoo)
toy_rds <- system.file('extdata', 'campylobacter.RDS', package = 'pagoo')
p <- load_pangenomeRDS(toy_rds)
もしくは、他のpan-genomeソフトウエアの結果を読み込ませる(上のデモデータを使う場合はスキップ)。
Roary(紹介)の結果を使う場合
roary_2_pagooを使ってroaryの結果をPagooのオブジェクトに変換する。roaryの出力のgene_presence_absence_csv以外にGFFファイルのパスも指定する必要がある。
gffs <- list.files(path = ".",
pattern = "[.]gff$",
full.names = TRUE)
gpa_csv <- "gene_presence_absence.csv"
library(pagoo)
p <- roary_2_pagoo(gene_presence_absence_csv = gpa_csv, gffs = gffs)
Pirate(紹介)の結果を使う場合
pirateスタイルのgene_presence_absence_csvをroaryスタイルに変換後、上のroaryと同じ手順でPagooのオブジェクトに変換する(4のセパレータに注意)。
#1 まずpiratesのスクリプトPIRATE_to_roary.plでroaryスタイルのgene_presence/absence.csvに変換する(#46)
PIRATE_to_roary.pl -i PIRATE.gene_families.ordered.tsv -o PIRATE_to_roary_gene_families.csv
#あとはroaryと同じように進める
#2 GFF (piratesが出力するmodified_gff/のGFFを使用しないとエラーになる)
gffs <- list.files(path = ".",
pattern = "[.]gff$",
full.names = TRUE)
#3 gene_presence/absence.csv
gpa_csv <- "PIRATE_to_roary_gene_families.csv"
#4(同一セル内でのパラログセパレータが";"であることを指定しないとエラーになる)
library(pagoo)
p <- roary_2_pagoo(gene_presence_absence_csv = gpa_csv, gffs = gffs, paralog_sep = ";")
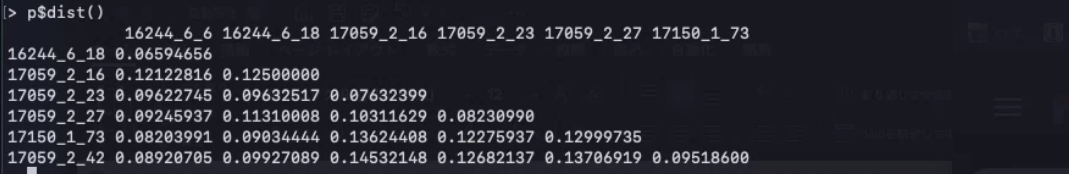
2,(link)を使った全ゲノムペア間のGene Abundance Distanceの計算(veganパッケージのvegdist()のラッパー)。デフォルトの距離法は Bray-Curtis距離(bray)。
p$dist()
#Jaccard距離に変更。このメソッドはpresence/absenceデータのみ必要とし、存在量を必要としない。引数 binary = TRUE をセットして使用する必要がある。
p$dist(method = "jaccard", binary = TRUE)
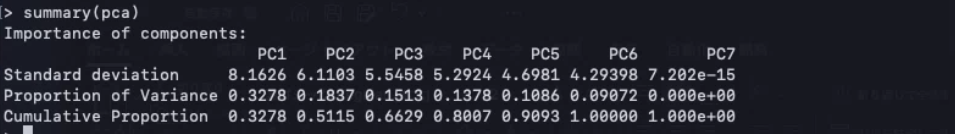
3、主成分分析
pca <- p$pan_pca()
summary(pca)
4、パイチャート
library(ggplot2)
library(patchwork) #複数のggplot2グラフを並べる
#PDF保存なら
pdf("PCA.pdf", width = 8, height = 6)
plot(pca)
dev.off()
#pie1
pdf("pie1.pdf", width = 8, height = 6)
p$gg_pie() + ggtitle("Default")
dev.off()
#pie1、pie2を並べる
pdf("pies.pdf", width = 8, height = 6)
pie1 <- p$gg_pie() + ggtitle("Default")
pie2 <- pie1 + ggtitle("Customized") + theme_bw(base_size = 15) + scale_fill_brewer(palette = "Blues")
pie1 + pie2
dev.off()
pie1
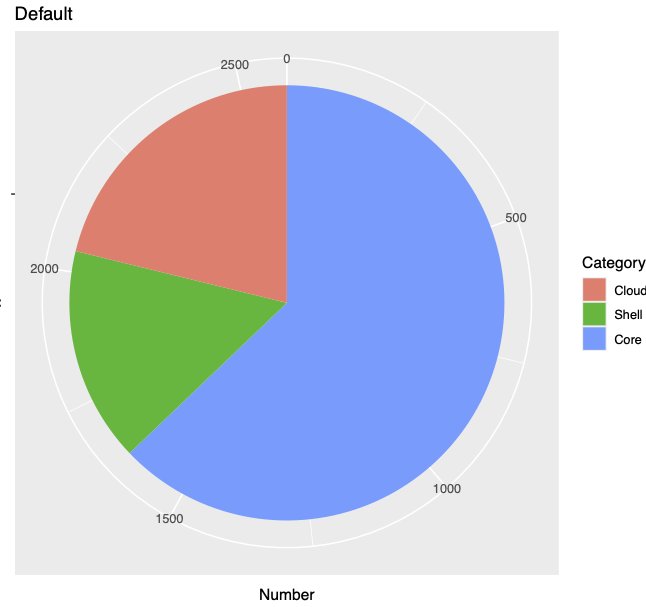
pie1 + pie2

5、Gene Frequency Bar plot
pdf("bar-plot.pdf", width = 8, height = 6)
p$gg_barplot()
dev.off()

均一な集団で比較するとU字状のプロットが得られる。内部ピークがあるなら、集団の異質性を示している可能性がある(Mikhail A. Moldovan and Mikhail S. Gelfand, 2018)。
6、Gene Presence/Absence BinMap
pdf("binmap.pdf", width = 8, height = 6)
p$gg_binmap()
dev.off()

ゲノムごとの遺伝子の有無を示すバイナリマップ
7、Rarefaction Curves
pdf("rarefaction_curves.pdf", width = 8, height = 6)
p$gg_curves()
dev.off()

(パンゲノムのサイズにフィットするためにべき乗分布が適用され、コアゲノムのサイズにフィットするために指数関数的減衰の関数が適用される。チュートリアルより)
カテゴリー別にポイントやファセットデータを追加したりカスタマイズできる。
pdf("rarefaction_curves_custom.pdf", width = 10, height = 6)
p$gg_curves() +
ggtitle("Pangenome and Coregenome curves") +
geom_point() +
facet_wrap(~Category, scales = 'free_y') +
theme_bw(base_size = 15) +
scale_color_brewer(palette = "Accent")
dev.off()

8、PCA Biplot - 遺伝子含量に基づいてゲノムの関連性を調べる
p$organisms
p$organisms

(このデモデータにはorganism、id、strain、year、country、host、source、accessionが含まれている)
プロット時、メタデータ"host"に基づいて色をつける。
pdf("pca-bbiplot.pdf", width = 8, height = 6)
p$gg_pca(colour = 'host', size = 4) +
theme_bw(base_size = 15) +
scale_color_brewer(palette = "Set2")
dev.off()

その他
- Pagooはバクテリアのパンゲノムを解析するソフトウェアフレームワークで、どのパンゲノム再構成ソフトウェアの出力でも使用できる。
- pagooの主なアイデアは、パンゲノムを再構築すると、すべての情報と基本的なメソッドがひとつのオブジェクトに組み込まれるということである。このオブジェクトにクエリーするには、Rで名前付きリストを使うように$シンボルを使うだけです。
- 遺伝学的、表現型、その他のメタデータを1つのオブジェクトまたはファイルに統合できる。
- 拡張性があり、他の微生物ゲノミクスパッケージと容易に相互作用する。
-
STAR Protocolsの論文では、より複雑な細菌集団に対してPagooを使って比較解析を行うためのプロトコルが提供されている。
引用
An object-oriented framework for evolutionary pangenome analysis
Ignacio Ferrés, and Gregorio Iraola
Cell Rep Methods. 2021 Sep 27; 1(5): 100085.
Protocol for post-processing of bacterial pangenome data using Pagoo pipeline
Ignacio Ferrés and Gregorio Iraola
STAR Protocols, Volume 2, Issue 4, 2021, 100802.
参考
https://github.com/SionBayliss/PIRATE/issues/46