パスウェイ解析(PA)は、機能的エンリッチメント解析としても知られている(引用)。パスウェイ解析ツールの目的は、ハイスループットテクノロジーから得られたデータを解析し、コントロールと比較してケースサンプルで変化している関連した遺伝子群(特定の機能を達成するために働く一連の遺伝子)を検出することにある。これを統計的に評価するなら、あるパスウェイの中で異なって発現する遺伝子の割合が、ランダムに予想される遺伝子の割合を上回る場合にそのパスウェイを報告する(引用)。パスウェイ解析を行うことで、重要かもしれないが孤立した個々の遺伝子の発現結果から、生物学的に意味のある結果を得ることができる。パスウェイ解析から、ユーザーは新たな仮説を立て、新しい実験を行い、その結果をさらに検証することができるようになる(引用)。
タンパク質相互作用を調べるSTRINGデータベースには、ユーザーのクエリを入力としてこのパスウェイ解析、すなわち機能的エンリッチメント解析を行い、その結果をweb上で閲覧する機能を持つ。しかし、STRINGで以前から利用できた機能的エンリッチメント解析では、ユーザーの元のファイルにはあったかもしれないfold changeなどの数値情報や、ほかの意味のあるランクは利用できなかった。入力タンパク質のランク付けは、入力リストが膨大である場合、どのようにランク付けされているかを知るためにも重要になる(引用)。STRINGは、バージョン11.0アップデートからこのランクベースのエンリッチメント検出アルゴリズム(機能クラススコアリング)に対応した。これは、各タンパク質や遺伝子にユーザーが入力した数値が付いている大規模な入力に対応するもので、統計的に有意なパスウェイを報告する実装となる(詳細はSTRINGの論文を読んで下さい)。この機能を試してみる。
STRING: functional protein association networksにアクセスし、Proteins with Values/Ranksを選択する。

実験全体のタンパク質リストを入力する。1列目には各タンパク質名やID、2列目には値(fold-change、log-pvalue、abundance)を入力する。ここではexample3を指定した。
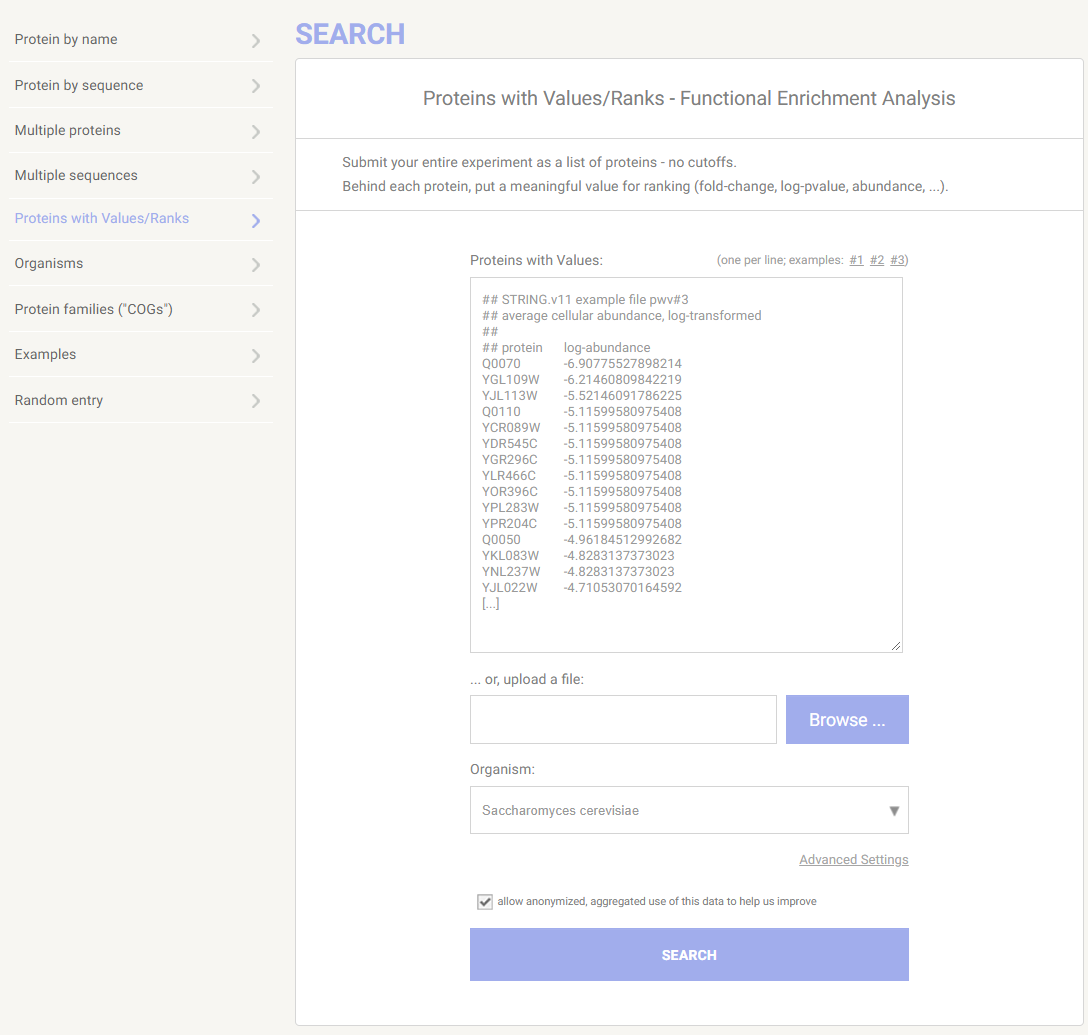
リストとしては見えていないが、3213クエリ入力されている。この例のように、利用可能なタンパク質ー値のペアの完全なリスト、理想的にはゲノム全体を使うのが推奨されている(引用)。
次のページでSTRING-dbの対応するプロテインへのマッピング結果が示される。マッピング結果に間違いがあればこのページで修正する。
出力例(example3 default setting)

GO、KEGG pathway、Wikipathway、Reactome、STRING-dbのPubmedテキストマイニング、Pfam、uniprotなどのランクベースのエンリッチメント解析結果がまとめられている。
enrichment socreの高い順にソートされている。

descriptionをクリックすると、検出された遺伝子の値と分布が表示される。enrichment socreの最も高いモジュールはintron関連のものだった。機能的モジュールの8遺伝子中3遺伝子が見つかっただけだが、その3遺伝子全ての発現が全遺伝子の発現分布の底付近に集中していた。

左端のGO termをクリックすると各データベースにジャンプする(GOならAmiGO2)。
pathway sizeをクリックすればSTRINGのネットワークで表示・分析できる。
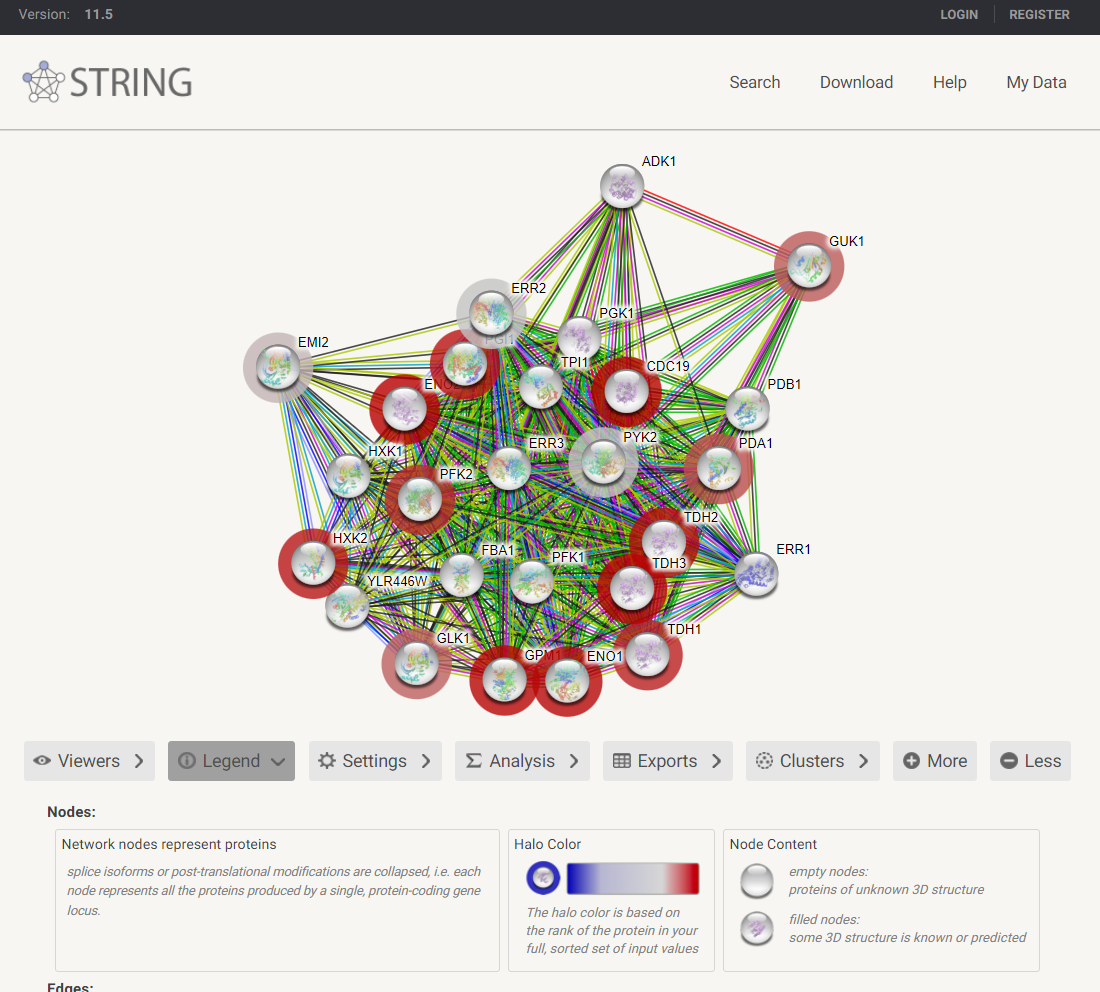
下の方には入力データの統計がまとめられている。

引用
The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets
Damian Szklarczyk, Annika L Gable, Katerina C Nastou, David Lyon, Rebecca Kirsch, Sampo Pyysalo, Nadezhda T Doncheva, Marc Legeay, Tao Fang, Peer Bork, Lars J Jensen, Christian von Mering
Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D605–D612
関連