DNAシーケンシングの技術進歩に伴い、バクテリアゲノムのショートリードによる全ゲノムアセンブリ(WGA)は、ごく一般的な作業となっている。ゲノムのアセンブリプロセスには絶対的な黄金律がなく、多くの異なるツールを組み合わせて一連のステップを実行する必要がある。しかし、最終的なアセンブリの品質は、常に入力データの品質と強く関連している。このことを念頭に置いて、本著者らはWGA-LPを開発した。このパッケージは、微生物解析のための最先端のプログラムと、サンプルとアセンブリ結果の両方の品質をチェックし改善するための新しいスクリプトを接続するものである。WGA-LPは、保守的な汚染除去アプローチにより、汚染されたリードの場合でも高品質なアセンブリを作成できることが示されている。WGA-LPはGitHub (https://github.com/redsnic/WGA-LP) とDocker Hub (https://hub.docker.com/r/redsnic/wgalp)で公開されている。ノード可視化のためのウェブアプリはshinyapps.io (https://redsnic.shinyapps.io/ContigCoverageVisualizer/)でホストされている。
除染手順は、3つのプログラムの呼び出しを含むカスタムスクリプトに基づいており、BWA mem (Li (2013)), Samtools, (Li et al. (2009)), Bazam (Sadedin and Oshlack (2019))の3つのプログラムの呼び出しを含むカスタムスクリプトをベースにしている。生リードと、標的生物用と汚染物質用の2セットのリファレンスを入力とする。この手順は、論文図1および補足資料に詳しく示されている。この除染方法は保守的であり、標的生物のリードが廃棄される確率を減らすことができる。このパイプラインはスタンドアローンでも、他のホールゲノムアセンブリプログラムと組み合わせても使用することができる。WGA-LPは、一般的なラップトップパソコンでも実行可能になっている。
Guide (supplementary materials)
Evaluate Node Coverage in bacterial WGA
https://redsnic.shinyapps.io/ContigCoverageVisualizer/
ガイドで丁寧に説明されています。ここでは流れだけ確認しておきます。
インストール
ubuntu18で配布されているdockerイメージをpullしてテストした。
依存
- bamtools: manage .bam files
- bazam: convert .bam files back to .fastq
- bracken: postprocess kraken2 reports to find contamination
- BWA: align .fastq files to a reference genome
- FastQC: evaluate .fastq quality
- kraken2: evaluate possible contaminations of the sequenced sample (minikraken db is required)
- mauve: program for multiple alignment, used to reorder contigs
- minia: a simple assembler for bacterial genomes
- prokka: annotate assembled genomes from bacteria
- samtools and plot-bamstats: manage .sam and .bam files, create reports
- SPAdes: a more complex assembler for bacterial genomes
- TrimmomaticPE: tool to clean .fastq reads
- checkM, merqury and quast: tools to evaluate WGA quality
#docker (hub)
docker pull redsnic/wgalp:latest
実行方法
チュートリアルではこちらのショートリードシークエンシングデータが使用されている。MiseqのペアエンドシークエンシングデータでPediococcus Acidilacticiの全ゲノムショットガンシークエンシングデータとなっている。予めデータをダウンロードしておく(grabseqs紹介)。
grabseqs sra -t 8 -m metadata.csv -o fastq_dir SRR15265000
WGA-LPのdocker imageをランする。
docker run --rm -itv $PWD:/root/shared --privileged --name wgalp redsnic/wgalp
立ち上がった。

1、品質トリミングと汚染チェック。fastqc、Trimmomatic、PEkraken2とbrackenが使用される。
cd /root/shared
wgalp trim --fastq-fwd SRR15265000_1.fastq.gz --fastq-rev SRR15265000_2.fastq.gz --kraken-db $kraken_db --output trimming_step
出力
trimming_step/

trimming_step/kraken/kraken.report
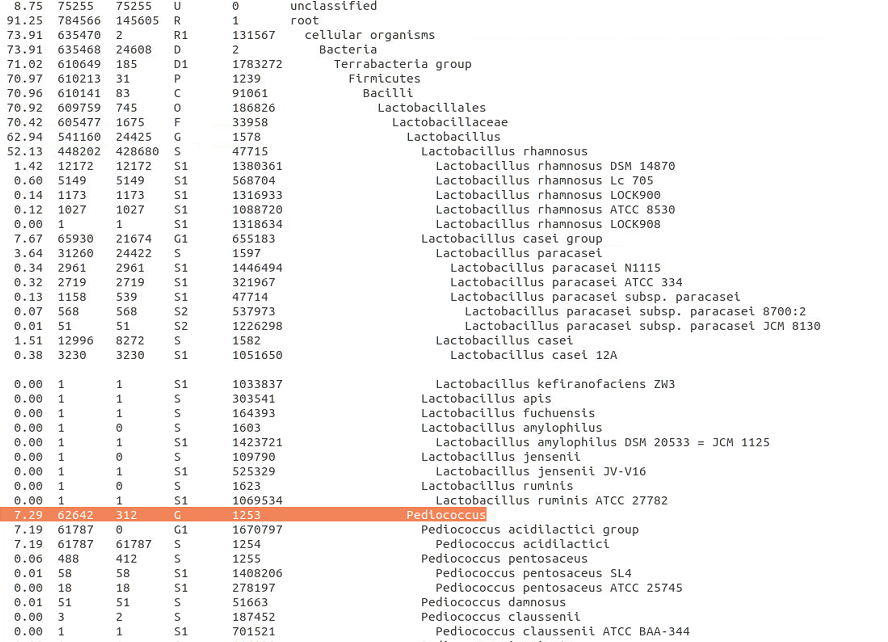
Lactobacillus属が62.94%と主要だが、下の方に行くと

Pediococcus属にも7.29%ヒットしている。単離細菌のシークエンシングとすると汚染が疑われる。
ここでは、このPediococcus属のゲノムを除くことにする。NCBI assemblyからPediococcus AcidilacticiとしてRefSeqに公開されているリファレンス配列全てをダウンロードする(リンク)。

tarボールを解凍後、中に入ってbwaのindexを作成しておく。チュートリアルにあるようにforループする。
cd genome_assemblies_genome_fasta/ncbi-genomes-2022-06-09/
for f in `ls *.fna`; do bwa index $f; done
リファレンスはLactobacillus rhamnosus とする。同様にダウンロードしてindexを作っておく(リンク)。ガイドではより少ないリファレンスが使用されている(ガイドの一番最後のページ参照)。
2、汚染配列を除く。入力されたリードは、汚染配列の各リファレンスに対して独立にマッピングされ、その後マージされ、徐々にフィルタリングされる。
cd /root/shared
wgalp decontaminate \
--fastq-fwd trimming_step/TrimmomaticPE/SRR15265000_1.trimmed.fastq \
--fastq-rev trimming_step/TrimmomaticPE/SRR15265000_2.trimmed.fastq \
--references ref/ncbi-genomes/*.fna \
--contaminants contami/ncbi-genomes/*.fna \
--output decontamination
decontaminantion/に結果は出力される。gzip圧縮されたfastqを使うとエラーになったので、解凍して使った。
decontamination/

3、結果を評価する。
wgalp understand-origin \
--fastq-fwd decontamination/decontaminated_fwd.fastq \
--fastq-rev decontamination/decontaminated_rev.fastq \
--kraken-db $kraken_db \
--output kraken_after_decontamination
kraken/

4、アセンブルする。 WGA-LPはSPAdes(とSPAdes-Plasmid)とMiniaアセンブラをネイティブにサポートし、wgalpアセンブルコマンドによる実行のためのインターフェースを提供している。
wgalp assemble \
--assembler SPAdes \
--fastq-fwd decontamination/decontaminated_fwd.fastq \
--fastq-rev decontamination/decontaminated_rev.fastq \
--output SPAdes
SPAdes/SPAdes/
5、生成されたノードの実際のカバレッジを確認するために、アセンブルされた配列にリードをバックアラインする。
wgalp check-coverage \
--fastq-fwd decontamination/decontaminated_fwd.fastq \
--fastq-rev decontamination/decontaminated_rev.fastq \
--contigs SPAdes/SPAdes/scaffolds.fasta \
--output coverage
wgalp check-coverageは、bwaとsamtools depthに依存し、各ノードのcoverageと長さの概要を作成する。

6、カバレッジ分布を視覚化する。
wgalp view-nodes \
--depth coverage/samtools_depth/aligned_to_scaffolds.depth \
--all \
--output coverage_plots
coverage_plots/
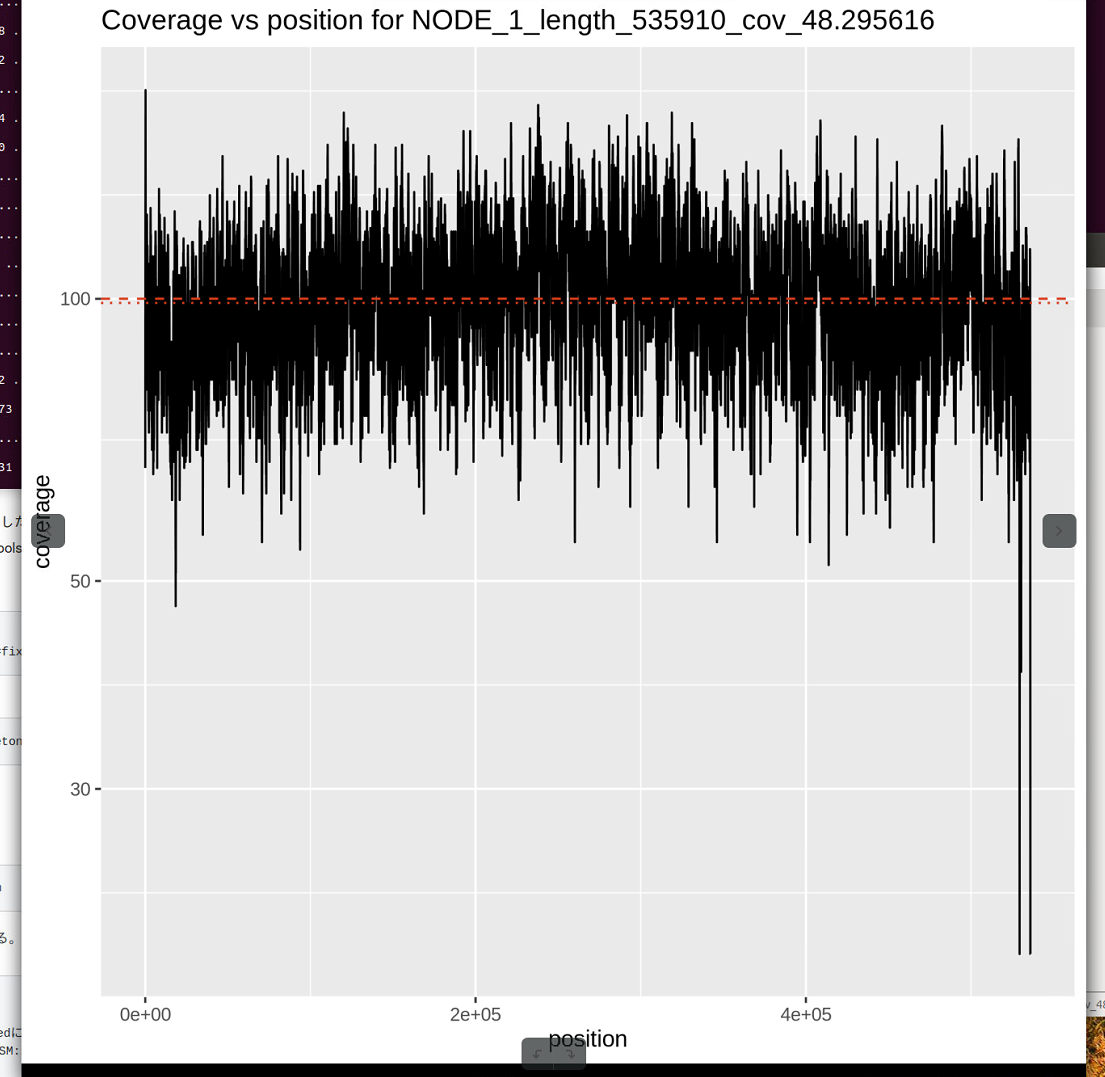
1つ開いてみる。
次のように、末端にバレッジのピークが含まれる配列もある。
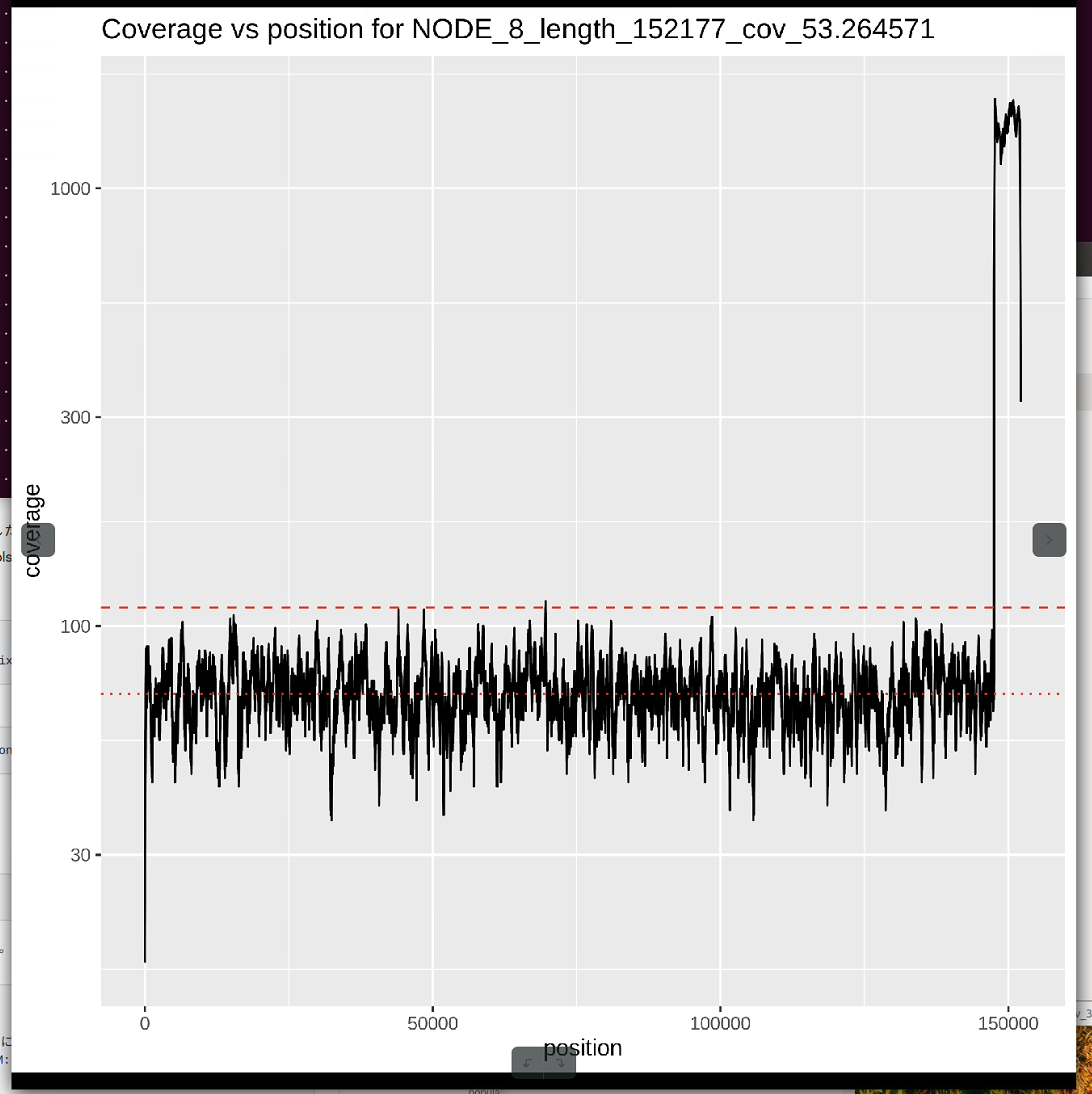
マニュアルでは、BLASTnサーチによって、末端の同様の末端のピークはバクテリオファージゲノムが細菌ゲノムに挿入されたことが原因である可能性が示唆されている。この配列の末端もBLASTn検索すればデプスが深い理由が推定できるかもしれない。
アセンブラが生成するノードの特徴をよりよく理解するためのウェブアプリも用意されている。
https://redsnic.shinyapps.io/ContigCoverageVisualizer/
ステップ5;wgalp check-coverageの.depth.summaryファイルをアップロードする。
視覚化例

縦軸は長さ(nt)、横軸はカバレッジ(リードデプス)。指定したカバレッジカットオフ以上のコンティグは青色のプロットになる。()の数値は左端のグラフに対応。
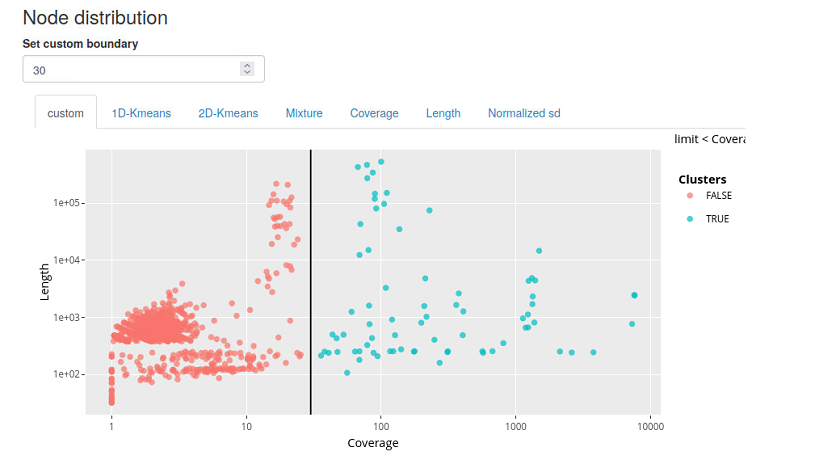
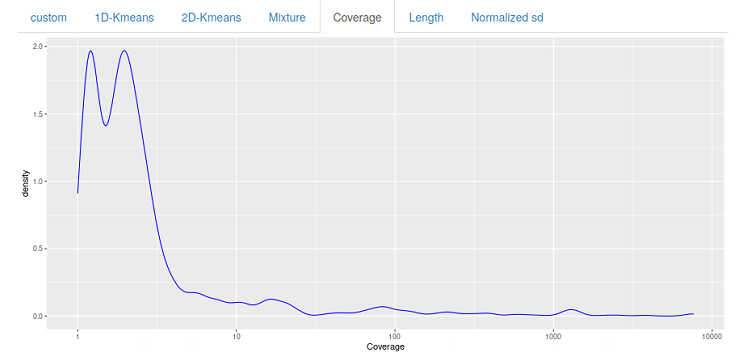
coverageヒストグラムプロットに変更。単離細菌のWGSでは通常は単峰性だが、2峰性のカバレッジプロットになっている。汚染が疑われる。
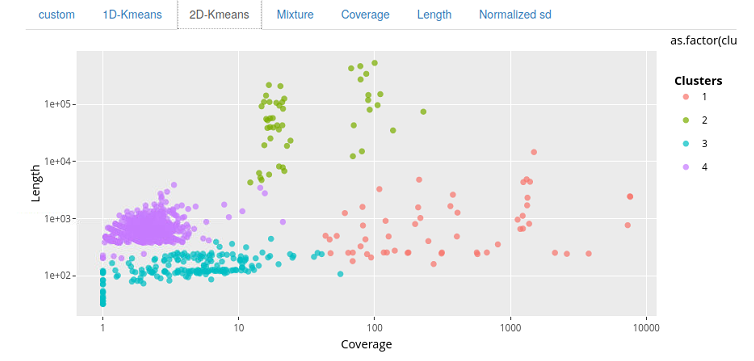
2D-KMeans-Clustering(色はクラスタ番号に対応)
コピーボタンを押すと、カットオフ以上のコンティグ名がコピーされる。ここではそれをselection30.txtとして保存した。
7、フィルタリングする。先ほどのselection30.txtを指定する。
wgalp filter-assembly \
--contigs SPAdes/SPAdes/scaffolds.fasta \
--selected-contigs selection30.txt \
--output filtered_contigs
filtered_contigs/

8、Kraken2を使って汚染物質のリードのアセンブリがあるかどうかをチェックする。
wgalp understand-origin \
--fasta filtered_contigs/filtered_contigs.fasta \
--kraken-db $kraken_db \
--output node_origin
node_origin/

マニュアルでは、分類がおかしいコンティグについてBLASTn検索している。
9、.fastaファイルを手動で編集するか、wgalp filterassembly --complement を使用することで、不要なノードを削除する。
wgalp reorder \
--contigs precise_filter/filtered_contigs.fasta \
--reference ../references/rhamnosus/LrhamnosusGGATCC.fasta \
--output reordering
10、プラスミドを抽出する。SPAdes plasmidかRecyclerを使う。Recyclerはwgalp plasmidコマンドで実行できる。
wgalp plasmid \
--fastq-fwd decontamination/decontaminated_fwd.fastq \
--fastq-rev decontamination/decontaminated_rev.fastq \
--contigs precise_filter/filtered_contigs.fasta \
--assembly-graph SPAdes/SPAdes/assembly_graph.fastg \
--kmer 127 \
--output recycler

このランでは2つの推定プラスミドが得られた。
11、QC。WGA-LPには、出来上がったアセンブリの品質テストを行うためのプログラム群が含まれている。これには、Quast,checkM、およびMerquryが含まれる。
wgalp quality \
--fastq-fwd decontamination/decontaminated_fwd.fastq \
--fastq-rev decontamination/decontaminated_rev.fastq \
--assembly reordering/mauve_reorder/alignment2/filtered_contigs.fasta \
--kmer-length 16 \
--output quality_control
quality_control/

12、アノテーション。WGA-LPには、NCBI準拠のアノテーションを作成するためProkkaが含まれている。
wgalp annotate \
--contigs reordering/mauve_reorder/alignment2/filtered_contigs.fasta \
--output annotation
annotation/
上のガイドの最後の方のページでは、パイプラインで使用されるフィルタリングがゲノムアセンブリ品質をどのように改善させるか、shovillなどと比較して示されています。興味がある方は確認して下さい。
引用
WGA-LP: a pipeline for whole genome assembly of contaminated reads
N Rossi, A Colautti, L Iacumin, C Piazza
Bioinformatics, Volume 38, Issue 3, 1 February 2022, Pages 846–848
関連