2021 5/18 ツイート追加
2021 11/11 論文引用
2022/03/10 help更新2023/10/16 インストール手順修正(python3 => python 3.10)
2024/08/22 webサーバーリンク追加
Baktaは、バクテリアのゲノムやプラスミドのローカルなアノテーションを迅速かつ標準化するためのツールである。dbxref-richやsORF-including annotationsを機械的に読めるJSONやバイオインフォマティクスの標準的なファイル形式で提供し、下流の自動解析を可能にしている。
Githubより
Baktaはバクテリアとプラスミドのみをアノテーションするために設計されました。
FAIRアノテーション FAIRの原則に則った標準的なアノテーションを提供するために、BaktaはUniProtのUniRef100とUniRef90のタンパク質クラスタ(FAIR -> DOI/DOI)をベースに、dbxrefs(GO, COG, EC)で強化された包括的なカスタムアノテーションデータベースを利用し、専門のニッチデータベースでアノテーションを行う。dbバージョンごとに、インポートされたすべての配列とアノテーションの包括的なログファイルを提供する。
FAIRの観点から、BaktaはRefSeq (WP_*), UniRef100 (UniRef100_*), UniParc (UPI*)へのデータベース相互参照(dbxref)でアノテーションされたMD5ダイジェストを介して、同一のタンパク質配列(IPS)を同定している。これにより、IPSでは、異なる遺伝子のアレルを監視し、比較分析を効率化することができる。また、これらの正確で安定した識別子を介して既存のCDSにマッピングすることができる、推定および仮説的なタンパク質配列の後付け(外部)アノテーションも可能である。現在、BaktaはUniParc、UniRef100、RefSeqからそれぞれ約1億9800万、約1億8500万、約1億5000万の異なるタンパク質配列を同定している。したがって、特定のゲノムについては、コストのかかるホモロジー検索を省略して、全CDSの99%までをこの方法で同定することができる。
ショートオープンリーディングフレーム 標準的なフィーチャータイプ(tRNA、tmRNA、rRNA、ncRNA遺伝子、ncRNAシス制御領域、CRISPR、CDS、oriC/V/T、gaps)に加えて、BaktaはProdigalのようなツールでは予測されない低分子タンパク質やショートオープンリーディングフレーム(sORF)を検出し、アノテーションを付ける。
Baktaは、典型的なバクテリアのゲノムをラップトップで10±5分、プラスミドを数秒/数分でアノテーションできる。
AMRやVF遺伝子など、関心の高い特定のタンパク質に対して高品質なアノテーションを提供するために、Baktaは様々なエキスパートアノテーションシステムを統合している。現在、BaktaではAMR遺伝子のアノテーションにNCBIのAMRFinderPlusを使用している。また、各配列に対して明確なカバレッジ、アイデンティティ、プライオリティーの値を持つ一般化されたタンパク質配列のエキスパートシステムは、現在VFDBとNCBIのBlastRulesで構成されている。
標準化された、分類群に依存しない、ハイスループットでローカルな方法でバクテリアのゲノムをアノテーションすることで、Baktaは、PGAPのような完全な機能を持つが計算負荷の高いパイプラインと、Prokkaのような迅速で高度にカスタマイズ可能なオフラインツールの間のニッチをターゲットにしている。実際、BaktaはProkkaに大きくインスパイアされており(Torsten Seemann氏に感謝)、多くのコマンドライン・オプションは、相互運用性とユーザーの利便性のために互換性がある。したがって、もしBaktaがあなたのニーズに合わなければ、ぜひProkkaを試してみてください。
2023/02/28
🦠🧬🖥️ Excited to announce Bakta v1.7.0 AND a major DB update!
— Oliver Schwengers 🇩🇪🇪🇺🇺🇦 (@oschwengers1) February 27, 2023
New Features:
- A lightweight DB version -> only 1.2 Gb (zipped)
- Gene symbol curation for CDS & operons
- A metagenome modehttps://t.co/RZsVqIfP2N
More information below 👇 (1/10)
2021 5/18
🦠🧬🖥️ Excited to share release v1.0 of Bakta: a new tool for rapid & standardized annotations of bacterial genomes & #plasmids https://t.co/HbZv6KhT4y more details 👇 1/n
— Oliver Schwengers (@oschwengers1) 2021年5月17日
インストール
condaで仮想環境を作ってテストした。また、オーサーのdocker imageもpullしてテストした。
mamba create -n bakta python=3.10 -y
conda activate bakta
mamba install -c conda-forge -c bioconda -c defaults bakta -y
#pip
python3 -m pip install --user bakta
#docker image (hub link)
docker pull oschwengers/bakta:latest
#singularity
singularity build bakta.sif docker://oschwengers/bakta:latest
singularity run bakta.sif --help
> bakta -h
usage: bakta [--db DB] [--min-contig-length MIN_CONTIG_LENGTH] [--prefix PREFIX] [--output OUTPUT] [--genus GENUS] [--species SPECIES] [--strain STRAIN] [--plasmid PLASMID] [--complete]
[--prodigal-tf PRODIGAL_TF] [--translation-table {11,4}] [--gram {+,-,?}] [--locus LOCUS] [--locus-tag LOCUS_TAG] [--keep-contig-headers] [--replicons REPLICONS] [--compliant]
[--proteins PROTEINS] [--skip-trna] [--skip-tmrna] [--skip-rrna] [--skip-ncrna] [--skip-ncrna-region] [--skip-crispr] [--skip-cds] [--skip-sorf] [--skip-gap] [--skip-ori] [--help]
[--verbose] [--threads THREADS] [--tmp-dir TMP_DIR] [--version]
<genome>
Rapid & standardized annotation of bacterial genomes, MAGs & plasmids
positional arguments:
<genome> Genome sequences in (zipped) fasta format
Input / Output:
--db DB, -d DB Database path (default = <bakta_path>/db). Can also be provided as BAKTA_DB environment variable.
--min-contig-length MIN_CONTIG_LENGTH, -m MIN_CONTIG_LENGTH
Minimum contig size (default = 1; 200 in compliant mode)
--prefix PREFIX, -p PREFIX
Prefix for output files
--output OUTPUT, -o OUTPUT
Output directory (default = current working directory)
Organism:
--genus GENUS Genus name
--species SPECIES Species name
--strain STRAIN Strain name
--plasmid PLASMID Plasmid name
Annotation:
--complete All sequences are complete replicons (chromosome/plasmid[s])
--prodigal-tf PRODIGAL_TF
Path to existing Prodigal training file to use for CDS prediction
--translation-table {11,4}
Translation table: 11/4 (default = 11)
--gram {+,-,?} Gram type for signal peptide predictions: +/-/? (default = ?)
--locus LOCUS Locus prefix (default = 'contig')
Locus tag prefix (default = autogenerated)
--keep-contig-headers
Keep original contig headers
--replicons REPLICONS, -r REPLICONS
Replicon information table (tsv/csv)
--compliant Force Genbank/ENA/DDJB compliance
--proteins PROTEINS Fasta file of trusted protein sequences for CDS annotation
Workflow:
--skip-trna Skip tRNA detection & annotation
--skip-tmrna Skip tmRNA detection & annotation
--skip-rrna Skip rRNA detection & annotation
--skip-ncrna Skip ncRNA detection & annotation
--skip-ncrna-region Skip ncRNA region detection & annotation
--skip-crispr Skip CRISPR array detection & annotation
--skip-cds Skip CDS detection & annotation
--skip-sorf Skip sORF detection & annotation
--skip-gap Skip gap detection & annotation
--skip-ori Skip oriC/oriT detection & annotation
General:
--help, -h Show this help message and exit
--verbose, -v Print verbose information
--threads THREADS, -t THREADS
Number of threads to use (default = number of available CPUs)
--tmp-dir TMP_DIR Location for temporary files (default = system dependent auto detection)
--version show program's version number and exit
Version: 1.3.3
DOI: 10.1099/mgen.0.000685
URL: github.com/oschwengers/bakta
Citation:
Schwengers O., Jelonek L., Dieckmann M. A., Beyvers S., Blom J., Goesmann A. (2021).
Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification.
Microbial Genomics, 7(11). https://doi.org/10.1099/mgen.0.000685
データベースの準備
ダウンロードする。圧縮状態で30GB近くあるので注意。
#2022/03/10更新
wget https://zenodo.org/record/5961398/files/db.tar.gz
tar -xzf db.tar.gz
rm db.tar.gz
amrfinder_update --force_update --database db/amrfinderplus-db/
ランの際はこのdb/を指定する。
=> 2022/03/10
データベースのダウンロードコマンドが使えるようになった。
#利用可能なDBのチェック
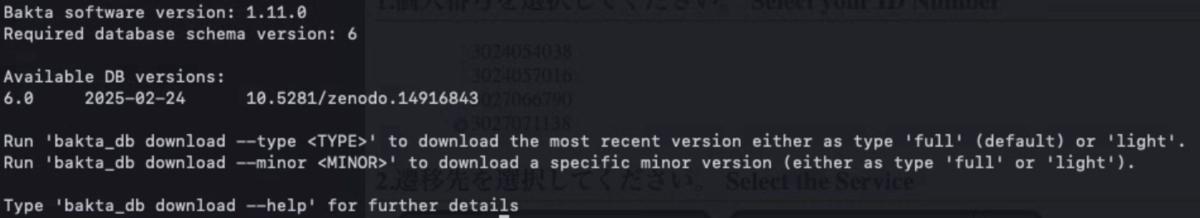
bakta_db list
$ bakta_db list
Required database schema version: 3
Available DB versions:
3.0 2021-08-05 10.5281/zenodo.5215743
3.1 2022-02-03 10.5281/zenodo.5961398
最新のDBをダウンロードする。
bakta_db download --output baktaDB_dir [light|full]
#あるいは手動でダウンロード(full)
wget https://zenodo.org/record/14916843/files/db.tar.xz
bakta_db install -i db.tar.xz
#aria2cで並列ダウンロード(8並列)
aria2c -x 8 -s 8 -c https://zenodo.org/record/14916843/files/db.tar.xz
bakta_db install -i db.tar.xz
#既存のDBをアップデート
bakta_db update --db baktaDB_dir --tmp-dir tmp
#パスを通す
export BAKTA_DB=baktaDB_dir
2025/05/14
実行方法
データベースとゲノムの(zipped)fastaファイルを指定する。
bakta --db db/ genome.fasta --threads 12
出力

アノテーションがつかなかったタンパク質配列はhypothetical proteinとして別ファイル出力される。
genome.gbff
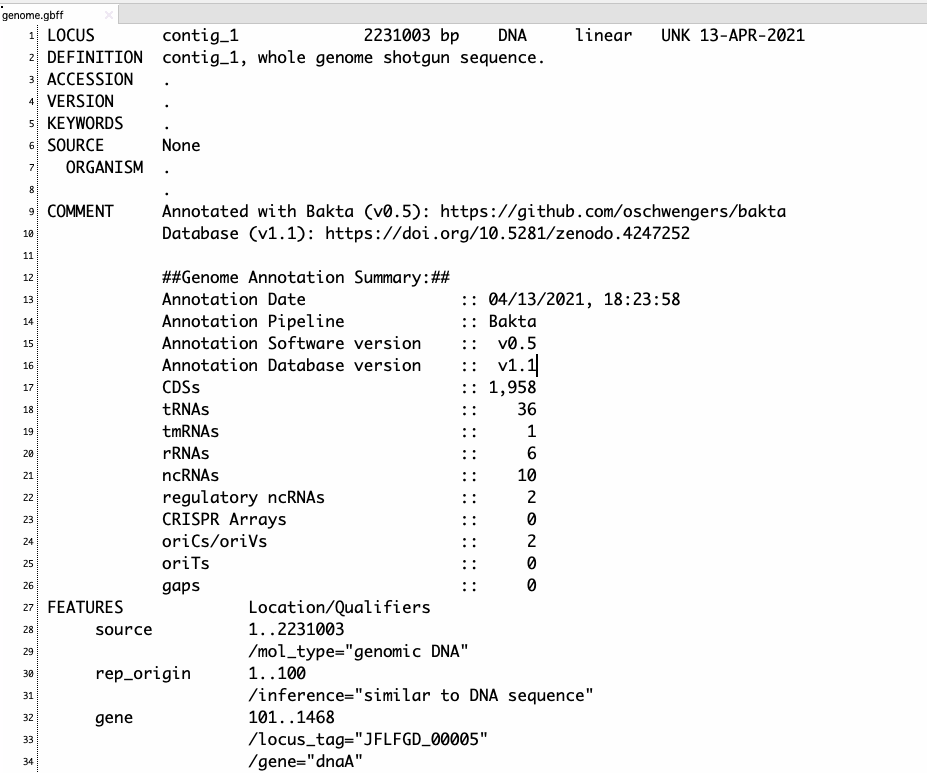
rRNAがtRNAとなっていたので修正した。
出力prefix指定、locus tagはlocusに指定、スレッド数は8。 verboseモード。
bakta --db db/ --verbose --output results/ --prefix outprefix --locus-tag locus --threads 8 genome.fna
出力

手持ちのタンパク質を使う場合はbakta_proteinsコマンドを使う。
bakta_proteins --db db/ --prefix test --output test --proteins special.faa --threads 8 input.fasta
追記
新しいバージョンではORFをplotした図も出力される。
ループ処理
mkdir protein
for file in `\find *fasta -maxdepth 1 -type f`; do
genome=${file}
folder=${file%.fasta}
bakta --db db/ --output ${folder} --locus-tag locus --threads 8 $genome
cp ${folder}/${file%.fasta}.faa protein/
done
その他
- webサーバー:
- レポジトリではready-to-goでNCBIやEMBLへゲノムとアノテーションを登録するためのサポートコマンドも紹介されている。
- collect-annotation-stats.py: ゲノムのコホートのアノテーション統計情報を収集し、凝縮したTSVを出力
- extract-region.py: 指定されたゲノム範囲内のゲノム特徴を抽出し、GFF3, Embl, Genbank, FAA, FFNとしてエクスポート
引用
Schwengers O., Goesmann A. (2021) Bakta: Rapid & standardized annotation of bacterial genomes & plasmids. GitHub https://github.com/oschwengers/bakta
2021/11/11
Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification
Oliver Schwengers, Lukas Jelonek, Marius Alfred Dieckmann, Sebastian Beyvers, Jochen Blom, Alexander Goesmann
Microb Genom. 2021 Nov;7(11)
関連