臨床サンプルのメタゲノムシークエンシングは、バイオサーベイランスにおける病原体の直接的な検出と特性解析に有望な手法である。菌株レベルでの分類学的解析は、バイオサーベイランスにおいて病原体の血清型を決定するために使用できる。Sigmaは、メタゲノム解析に基づく参照ゲノムを用いた病原体の株レベルでの同定と定量化のために開発された。Sigmaは、同定されたゲノムの仮説検定とその相対量の信頼区間推定を含む、推論の統計的不確実性を定量化する。Sigmaは、メタゲノミックリードを最も可能性の高い参照ゲノムに割り当てることにより、菌株のバリアントコーリングを可能にする。アルゴリズムの性能は、模擬コミュニティとスパイクイン病原体株を含む糞便サンプルを用いて評価した。SigmaはC++で実装され、ソースコードとバイナリは http://sigma.omicsbio.org (リンク切れ)で自由に入手できる。
バイオサーベイランスではさらに、検出された病原体の一塩基多型(SNP)に関する情報が、異なる変異体集団を区別するために必要となる。分離株ゲノムシーケンスでは、すべてのリードを参照ゲノムにマッピングすることで、バリアントの同定は容易である。しかし、メタゲノムシーケンスでは、すべてのメタゲノムリードを単純に参照ゲノムにマッピングすることは難しい。なぜなら、アラインメントされたリードの一部は異なる微生物に由来する可能性があり、偽の変異を引き起こす可能性があるからである。Sigmaはこの問題を解決するために、確率モデルに基づき、各リードを最も可能性の高い由来ゲノムに割り当てる。これによってSAMToolsのようなバリアントコーリングソフトウェアによる正確なバリアントコーリングが可能になる。
Manual
https://github.com/BioHPC/Sigma/blob/main/SIGMA_UserManual_1.1.0.pdf
インストール
g++ (gcc version 4.7.2)でコンパイルしたビルド済みバイナリも含めて配布されている。make cleanせずそのまま使用した。
git clone https://github.com/BioHPC/Sigma.git
cd Sigma/bin/
chmod +x sigma*
export PATH=$PWD:$PATH
$ ./sigma-index-genomes -h
[Usage]
sigma-index-genomes [options] -c <config file path> -w <working directory>
[Inputs]
1. config file path (default: sigma_config.cfg)
- if config file is not specified, the program will search it in the working directory
- include bowtie search options and more
2. working directory (default: current running directory)
- if working_directory is not specified, the program will work in the current directory
- results will be generated in working directory
[Options]
-h/--help
-v/--version
-p/--multi-processes <int> # number of multi-processes (default: 1)
[Outputs]
Bowtie2 index files for each genome will be generated in the genome directory
> ./sigma-align-reads -h
[Usage]
sigma-align-reads [options] -c <config file path> -w <working directory>
[Inputs]
1. config file path (default: sigma_config.cfg)
- if config file is not specified, the program will search it in the working directory
- include bowtie search options and more
2. working directory (default: current running directory)
- if working_directory is not specified, the program will work in the current directory
- results will be generated in working directory
[Options]
-h/--help
-v/--version
-p/--multi-processes <int> # number of multi-processes (default: 1)
[Outputs]
bam format aligned results are reported in each output genome directory
> ./sigma-build-model -h
[Usage]
sigma-build-model [options] -c <config file path> -w <working directory>
[Inputs]
1. config file path (default: sigma_config.cfg)
- if config file is not specified, the program will search it in the working directory
2. working directory (default: current running directory)
- if working_directory is not specified, the program will work in the current directory
- results will be generated in working directory
[Options]
-h/--help
-v/--version
[Outputs]
Q matrix file: sigma_out.qmatrix.txt will be generated in the working directory.
> ./sigma-solve-model -h
[Usage]
sigma-solve-model [options] -c <config file path> -w <working directory>
[Inputs]
1. config file path (default: sigma_config.cfg)
- if config file is not specified, the program will search it in the working directory
2. working directory (default: current running directory)
- if working_directory is not specified, the program will work in the current directory
- results will be generated in the working directory
[Options]
-h/--help
-v/--version
-i/--input-qmatrix <string> # provide q-matrix filename directry
-t/--multi-threads <int> # number of threads (default: 1)
[Outputs]
sigma_out.gvector.txt
sigma_out.gvector.html
sigma_out.ipopt.txt
実行方法
使用するにはconfigurationファイルを設定する。この内容に従って実行される。
sigma_config.cfg
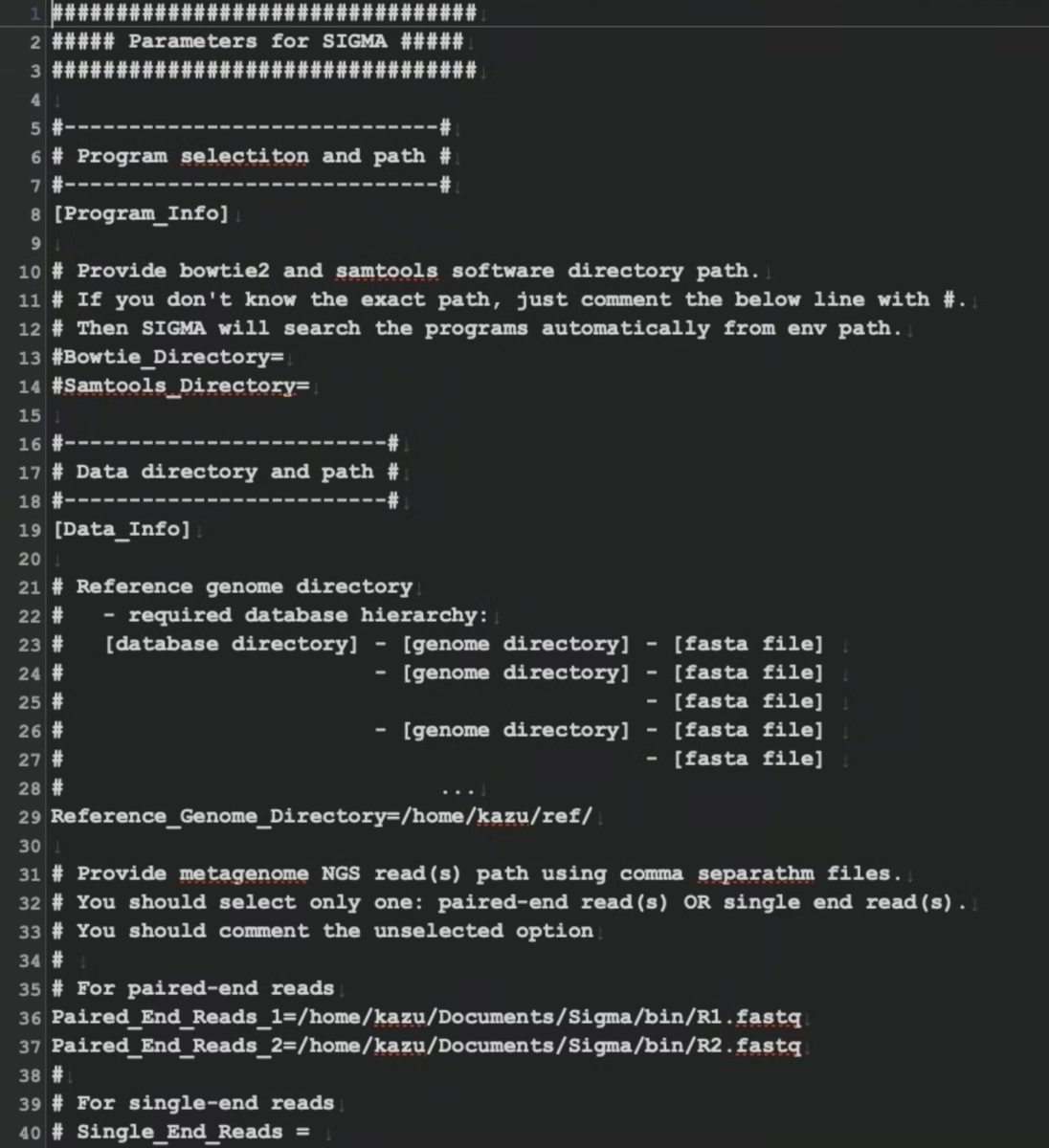
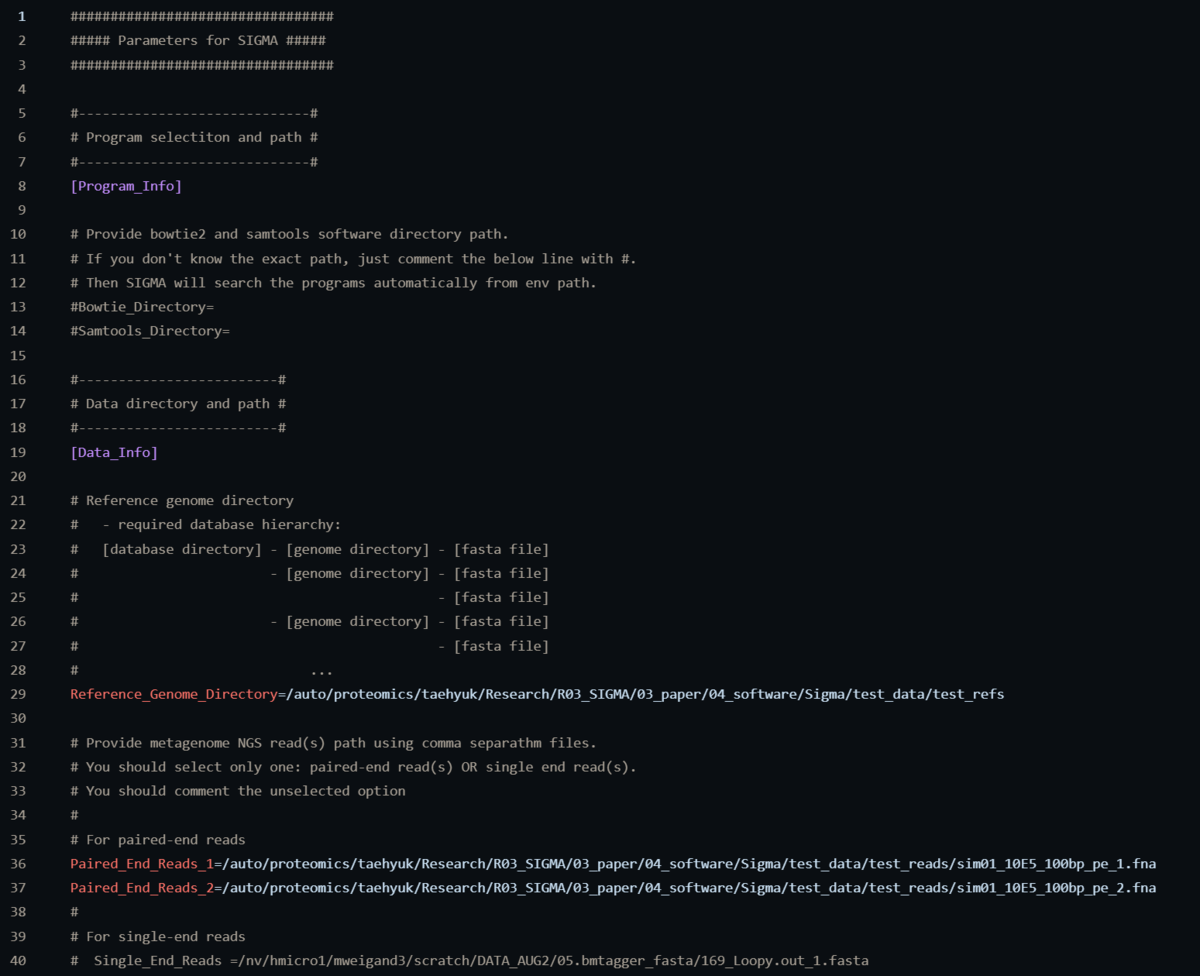
リファレンスゲノムディレクトリは"Reference_Genome_Directory="で親ディレクトリのパスを指定する。実際には上の画像で説明されているように、親ディレクトリの中野のサブディレクトリにfastaファイル(.fasta)を配置する。複数ファイル含めてもOKだが、1つのグループとして扱われる点に注意。
fastqファイルのパスは”Paird_End_Reads=”で指定する。ほか、bowtie2などのパラメータなどを指定する(デフォルトでも実行可能)。生のfastqかfastaを指定する(圧縮非対応)。
testの例
Sigma/bin/test_config.cfg
Sigma/bin/test_config.cfg at main · BioHPC/Sigma · GitHub
実行方法
1,indexing
sigma-index-genomes -c sigma_config.cfg -p 20
- -p number of multi-processes (default: 1)
2、mapping(MPIも利用できる)
sigma-align-reads -c sigma_config.cfg -p 20
- -p number of multi-processes (default: 1)
結果はsigma_alignments_output/下にゲノムのサブディレクトリごとに出力される。
3、モデル推定
sigma-build-model -c sigma_config.cfg -w .

sigma_out.qmatrix.txtが出力される。
4、菌株推定
sigma-solve-model -c sigma_config.cfg -w . -t 20
- -t/--multi-threads <int> # number of threads (default: 1)

出力
sigma_out.html


論文より
- 既存の分類学的解析アルゴリズムは一般的に、その結果の統計的信頼性評価を提供していない。病原体の存在に関するアルゴリズムの推論に基づくバイオサーベイランスの決定を、そのような推論の不確実性を知らずに行うことや、ゲノムの推定相対存在量の信頼区間を知らずに行うことは困難である。ゲノムの同定と定量化後、Sigmaはその結果の統計的信頼性を厳密に評価することができる。尤度比検定に基づく仮説検定により、関連ゲノムの検出に関するP値を算出し、ブートストラップ法を用いて、同定されたゲノムの相対存在量の信頼区間を推定する。
引用
Sigma: Strain-level inference of genomes from metagenomic analysis for biosurveillance Open Access
Tae-Hyuk Ahn , Juanjuan Chai , Chongle Pan Author Notes
Bioinformatics, Volume 31, Issue 2, January 2015, Pages 170–177
関連