ロングリードシーケンス技術の急速な発展により、個体完全長ゲノムの時代が近づいている。本著者らは、様々な全ゲノムアラインメント(WGA)フォーマットをサポートし、アラインメントの変換、処理、評価、可視化のための実用的なツールを提供する、クロスプラットフォームの超高速ツールキットであるwgatoolsを開発した。
wgatoolsは様々なフォーマットをサポートし、アラインメントの処理、フィルタリング、統計的評価、アラインメントに基づいたバリアントコーリングの実行、ローカルおよびゲノムワイドのアラインメントの可視化ができる。wgatoolsは、MITオープンソースライセンスの下、フリーソフトウェアとして公開されており、そのソースコードは、https://github.com/wjwei-handsome/wgatools、https://zenodo.org/records/14882797にある。
インストール
ubuntu24にcondaを使ってインストールした。
#conda
mamba create -n wgatools -y
conda activate wgatools
mamba install wgatools -c bioconda -y
#build from source
git clone https://github.com/wjwei-handsome/wgatools.git
cd wgatools
cargo build --release
> wgatools
wgatools -- a cross-platform and ultrafast toolkit for Whole Genome Alignment Files manipulation
Version: 0.1.0
Authors: Wenjie Wei <wjwei9908@gmail.com>
Usage: wgatools [OPTIONS] <COMMAND>
Commands:
maf2paf Convert MAF format to PAF format [aliases: m2p]
maf2chain Convert MAF format to Chain format [aliases: m2c]
paf2maf Convert PAF format to MAF format [aliases: p2m]
paf2chain Convert PAF format to Chain format [aliases: p2c]
chain2maf Convert Chain format to MAF format [aliases: c2m]
chain2paf Convert Chain format to PAF format [aliases: c2p]
maf-index Build index for MAF file [aliases: mi]
maf-ext Extract specific region from MAF file with index [aliases: me]
chunk Chunk MAF file by length [aliases: ch]
call Call Variants from MAF/PAF file [aliases: c]
tview View MAF file in terminal [aliases: tv]
stat Statistics for Alignment file [aliases: st]
dotplot Plot dotplot for Alignment file [aliases: dp]
filter Filter records for Alignment file [aliases: fl]
rename Rename MAF records with prefix [aliases: rn]
maf2sam DEV: maf2sam [aliases: m2s]
pafcov Calculate coverage for PAF file [aliases: pc]
pafpseudo Generate pesudo-maf for divergence analysis from PAF file [aliases: pp]
gen-completion Generate completion script for shell [aliases: gc]
validate Validate and fix query&target position in PAF file by CIGAR
Examples:
wgatools validate wrong.paf // output report to STDOUT
wgatools validate wrong.paf -f happy.paf -o fix.report [aliases: vf]
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help (see more with '--help')
-V, --version Print version
GLOBAL:
-o, --outfile <OUTFILE> Output file ("-" for stdout), file name ending in .gz/.bz2/.xz will be compressed automatically [default: -]
-r, --rewrite Bool, if rewrite output file [default: false]
-t, --threads <THREADS> Threads, default 1 [default: 1]
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace, defalut: Warn]
オートコンプリート設定(Fishシェルでタブ補完(コマンド候補表示)が機能するようにする)
mkdir -p ~/.config/fish/completions/
wgatools gen-completion --shell fish > ~/.config/fish/completions/wgatools.fish

実行方法
1,フォーマット変換
MAF => PAF (Multiple Alignment Format => Pairwise mApping Format)
git clone https://github.com/wjwei-handsome/wgatools.git
cd wgatools/test/
wgatools maf2paf test.maf > test.paf

出力例
> cat test.paf
![]()
PAF => MAF
クエリとターゲットのfasta配列(.fa, .fa.gz)を指定する必要がある。
wgatools paf2maf test.paf --target target.fa --query query.fa > test.maf
PAF => CHAIN
wgatools paf2chain test.paf > test.chain
MAF => CHAIN
wgatools maf2chain test.maf > test.chain
CHAIN => PAF
wgatools chain2paf test.chain > test.paf
CHAIN => MAF
wgatools chain2maf test.chain --target target.fa --query query.fa > test.maf
2、dotplot: PAF|MAF|CHAIN形式のペアワイズアラインメントのDot plotによる視覚化
wgatools dotplot -f paf test/testdotplot.paf > out.html

出力例

一致(マッチ)、挿入、欠失など、アラインメントの詳細を掴み、ローカルアラインメントをより良く観察するのに役立つ。デフォルトでは、50bpより小さいINDELは隣接するマッチとマージされる。パラメータ -l, --length で閾値を指定できる(レポジトリより)。
3、tview: コマンドラインでのアラインメント視覚化
#indexが必要
wgatools maf-index test.maf
#tviewコマンドでmaf指定
wgatools tview test.maf

十字キーの左右でスクロールできる。

gを押すとナビゲーションウィンドウが表示され、左側にシーケンス名、右側に選択したシーケンスの任意のインターバルが表示される。
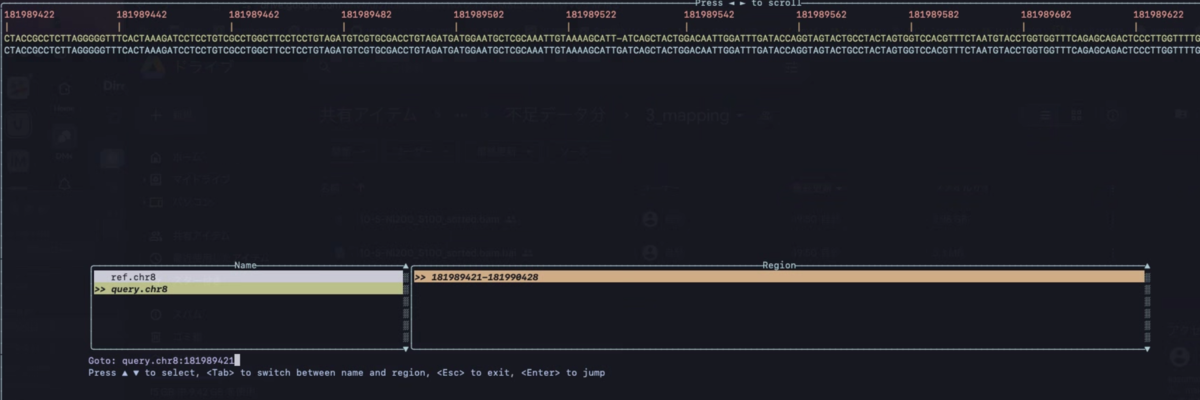
Tabを押すと左右の選択ウィンドウが切り替わり、また▲▼を押すとシーケンスとインターバルを選択できる。Enterを押してジャンプし、Escでナビゲーション・ウィンドウを閉じる。
4,stat: MAF|PAF|CHAINの統計
#MAF
wgatools stat test.maf
#PAF
wgatools stat -f paf test.paf
#CHAIN
wgatools stat -f chain test.chain
5, validate: PAFファイルを検証する。クエリーとターゲットの位置が違っていたり、CIGAR文字列が配列と不一致だったりとPAFファイルが正しくない場合、検証・修正に使う。
wgatools validate wrong.paf
#修復したpafを書き出す
wgatools validate wrong.paf -f happy.paf

出力例
> wgatools validate test.paf

6,call: MAF形式は各塩基のアラインメントを完全に記録しているため、バリアントコールに使用できる。
#defaultではSNPs、small indelはcallしない。SVのみコールする
wgatools call test.maf
#SNPsもコールするには”-s” と ”-l 0”をつける
wgatools call test.maf -s -l0
#PAFからダイレクトにバリアントコールするには、ターゲットとクエリの配列を指定する
wgatools call test/test.paf -s -l0 --target target.fa --query query.fa -f paf

出力例
> wgatools call test.maf -s -l0

#PAFからダイレクトにバリアントコールするには、ターゲットとクエリの配列を指定する
wgatools call test/test.paf -s -l0 --target target.fa --query query.fa -f paf
7, chunk : 巨大なMAFファイルを分割する。
wgatools chunk -l 100 test.maf -o chunked.maf

出力例
> wgatools chunk -l 100 test.maf -o chunked.maf

8,filter: ブロックサイズやクエリサイズ、アラインメントサイズでMAFをフィルタリングする。
wgatools filter test.maf -q 1000000 > filt.maf

9,rename: MAFのクエリとターゲットの配列名をリネームする。例えばrefとqueryの染色体名が同じchr1になっている場合など、配列名に接頭辞をつけてリネームする。
wgatools rename --prefixs REF.,QUERY. input.maf > rename.maf

出力例
> wgatools rename --prefixs REF.,QUERY. test.maf

10, pafcov: 複数のゲノムのアライメント結果がある場合にアライメントカバレッジを計算する。wfmash出力に最適化されている。BED3形式+4フィールド目にカバレッジ、のフォーマットで出力される。
wgatools pafcov all.paf > all.cov.beds

11、 pafpseudo: all versus all PAFから擬似MAFを生成する
wgatools pafpseudo -f all.fa.gz all.paf -o out_dir -t 10

引用
Wgatools: an ultrafast toolkit for manipulating whole genome alignments
Wenjie Wei, Songtao Gui, Jian Yang, Erik Garrison, Jianbing Yan, Hai-Jun Liu Author Notes
Bioinformatics, Published: 27 March 2025 Article history
関連