次世代シーケンサーによる実験では、データの探索的解析、トレンドの解釈、ターゲット/候補の特定、結果のわかりやすい直感的な可視化などが大きな課題となってる。これらの課題は、利用可能な解析ツールの大半がプログラミングスキルを必要とするため、コンピュータコードを書いた経験のない研究者にとってさらに高いハードルとなっている。また、計算生物学の専門家であっても、標準的な結果を得るためには、効率的で再現性のあるシステムが必要である。
著者らは、Pythonベースのモジュール式RNAシーケンスデータ解析ソフトウェアであるRNAlysisを開発した。RNAlysisは、生のFASTQファイルから、探索的データ解析、データの可視化、クラスタリング解析、遺伝子セットのエンリッチメント解析まで、研究課題に応じてカスタマイズした解析パイプラインを構築することが可能である。RNAlysisは親しみやすいグラフィカルユーザーインターフェースを提供し、研究者はコードを書かずにデータを分析することができる。線虫を用いた様々な研究からのRNAデータを解析することにより、RNAlysisの使用法を実証している。このソフトウェアは、どのような生物から得られたデータにも同様に適用できる。
RNAlysisは、様々な生物学的問題の調査に適しており、研究者はより正確で再現性のある包括的なバイオインフォマティクス解析を実行することができるようになります。また、多様なツール、拡張性、自動化、解析間の標準化を提供するため、経験豊富なバイオインフォマティシャンの解析をより堅牢かつ効率的にすることができる。
Documentation
https://guyteichman.github.io/RNAlysis/build/index.html
チュートリアル;時系列データセットとGEOから公開されている3つのデータセットを使った発現変動解析
https://guyteichman.github.io/RNAlysis/build/tutorial.html#analysis-2-differential-expression-data
RNAlysisでできる事
- 遺伝子発現マトリックス、差分発現表、フォールドチェンジデータ、表形式データ全般のフィルタリング
- 遺伝子発現マトリックスの正規化
- シーケンシングデータを可視化し、探索し、記述する
- クラスタリング解析と次元削減により、サンプル発現プロファイル間のグローバルな関係を発見する
- 解析パイプラインの作成と共有
- あらかじめ設定されたGene Ontology用語/KEGGパスウェイ、または使用済みの定義された属性を用いてエンリッチメント解析を実行する
- テストセットと遺伝子リストの代わりに、単一のランク付けされた遺伝子リストでエンリッチメント解析を実行する
リードカウントからの下流解析に加え、fastqを指定して定量から行うこともできます。実際の使い方について学ぶ場合、下流解析はチュートリアル1,fastqからの解析はチュートリアル2を読んでください。
インストール
macとwindowsに対応したスタンドアロンアプリはリリースからダウンロードできる。ここでは両OSのスタンドアロンアプリを試した(windows11もしくはMacstudioでMonterey使用)。
依存(All of RNAlysis’s dependencies can be installed automatically via PyPI)
numpy
pandas
scipy
matplotlib
numba
requests
scikit-learn
scikit-learn-extra
hdbscan
seaborn
statsmodels
joblib
tqdm
appdirs
grid_strategy
pyyaml
UpSetPlot
matplotlib-venn
xlmhg
pairwisedist
typing_extensions
PyQt5
qdarkstyle
defusedxml
cutadapt
aiohttp
aiodns
aiolimiter
Brotli
#PyPiからも導入可能
pip install RNAlysis
#起動
rnalysis-gui
実行方法
ダウンロードしたRNAlysis-3.6.2_macos.zipを解凍する。生じたRNAlysisを叩いて1分ほど待つと起動する。
起動したところ

実行を許可後しばらく待つ。

数分後に起動した。ガイドパネルが表示されており、動画で基本的な使い方を学ぶことができる。

ここでは”A-to-Z tutorial”で説明されているチュートリアルに従って進める。
WormBaseで公開されているデータ(direct link)
このデータセットでは、線虫の発生段階における各遺伝子の発現量の中央値が記載されている(文献のRNA-seq実験で「control」ラベルになっているデータ)。上のリンクでは中央値以外のカラムは削られている。チュートリアルには元のデータのリンクもある。
elegans_developmental_stages

各発達段階の発現量の中央値が記載されている。
LoadをクリックしてTSVファイルを読み込む。

データタイプをOtherからCount matrixに変更する。

変更することで、カウント行列形式のデータセットに特化した分析手法を使用できるようになる。
ウィンドウメニューが部分的に変わり、テーブルが事前に正規化されているかどうかを指定できるようになる。公開されているデータは既にFPKM正規化されているのでTrueに切り替える(緑色ボタン)。

startボタンをクリックすると表がプレビューされる。

表の内容(テーブル名、テーブルタイプ、行数、列数)の簡単な要約が表示されている。
表全体を見たい場合は「View full table」ボタンをクリックする。新しいウィンドウで表を確認できる。

中央右のsave tableをクリックすると、表をCSVかTSV形式で新しいファイルに保存することができる。書き出しは分析中のどの時点でも可能になっている。

全てのサンプルで発現していない、あるいは発現量が少ない遺伝子をフィルターで除外する。これらの遺伝子の発現の誤差は比較的大きく、これらの遺伝子は解析に有用なシグナルではなく、ノイズを加える可能性が高いため(チュートリアルより)。

"Filter genes with low expression in all columns "は、全ての列で発現レベルがXより低い遺伝子をフィルタリングする。つまり、少なくとも1つの条件下では閾値以上の発現をしている行は保持される。関数が何をするのかわからない場合は、関数名の横にある青いクエスチョンマークボタンをクリックする。Documentで説明を読むことができる。
Filter genes with low expression in all columnsを選択後、出てきたthreshouldメニューの値を選択する。

新しいスイッチボタンが2つ表示されている。oppositeスイッチをTrueに切り替えると、フィルタリング効果が反転する(閾値以下が残る)。低発現遺伝子以外がフィルタリングされることになる。
inplaceスイッチをTrueに切り替えると、フィルタリングされていないテーブルが保持され、新しいタブでフィルタリング操作を適用するかを選択することができる(デフォルトでTrue)。この機能は、複数のフィルタリングアプローチを並行して試したい場合や、フィルタリングされていないテーブルを後で使用する必要がある場合などに便利である(チュートリアルより)。どのような方法を選んでも、フィルタリング結果を明示的に保存しない限り、読み込んだ元のファイルがフィルタリングによって変更されることはない。適用したフィルタリング操作はボタンをクリックするだけで元に戻すことができる。
ここでは閾値を50にする。テーブルのすべての列で50 FPKM未満の遺伝子行がフィルタリングされる。

Applyをクリックするとフィルタリングが適用される。上の画像は適用後のもので、右側に履歴パネルが追加されている。この履歴をクリックすると、その時の表に戻すことができる。
履歴パネルのemptyをクリックした。表が最初の数値に戻った。

履歴をクリックして表を戻したりapplyすると下のlogパネルに結果が表示される。logパネルには遺伝子数がどれくらい変化したか書かれているので参考になる。
履歴パネルを閉じた時は上の白い帯部分を右クリックすると再表示できる。

Filterには、発現レベルによる遺伝子のフィルタリング、倍率変動の方向と大きさ、統計的有意性、欠損データのある行の除去など、さまざまなフィルタリング機能がある。

Filterは複数かけることができる。
各関数の詳細はDocumentの関数のセクションで説明されている。
https://guyteichman.github.io/RNAlysis/build/rnalysis.filtering.CountFilter.filter_low_reads.html
Normalizeボタンでは正規化方法を選択できる。今回は不要。

使い方はFilterと同じです。方法を選択後、出てきたパネルを操作し、最後にapplyします。
解析に戻る。
主成分分析を行う。visualizeから選択する。

https://guyteichman.github.io/RNAlysis/build/rnalysis.filtering.CountFilter.pca.html
パラメータを選択後、Applyをクリック。デフォルトではべき乗変換されてから主成分分析が適用される(べき乗変換を適用することで、歪み、平均-分散依存性、極値といったカウントデータの望ましくない特性を最小化する)。

主成分1-3の間の全ペアワイズでのグラフが生成される。グラフの数はn_componentsパラメータの値から変更できる(画像は3の時)。図はサイズを変更したり保存することができる。

主成分1-2がデータセットの分散の75%を説明している。
発生段階の遺伝子発現の類似性を調べるためにPair-plotを使う。
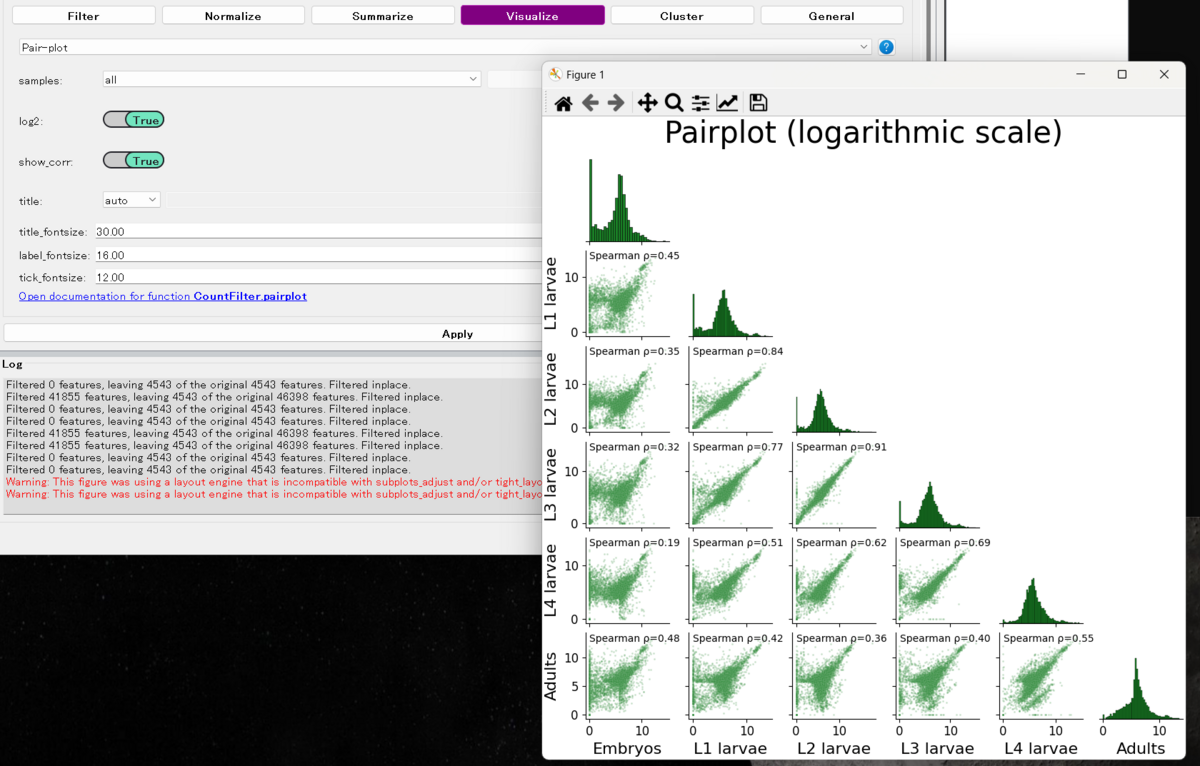
Pair-plot機能では実験条件ごとにサンプルをグループ化して調べることができる。log2変換のオプションもある。
胚の状態が他の条件と最も異なっているように見え、連続した発達段階は互いにかなり相関しているように見える(マニュアルより)。
特定の遺伝子の発現を見るにはplot expression of specific genesを選択する。

https://guyteichman.github.io/RNAlysis/build/rnalysis.filtering.CountFilter.plot_expression.html
set inputをクリックして遺伝子を選択する。出てきたパネルでadd fieldをクリック、WBGene00003864とWBGene00004804を入力する。Doneで閉じる。

遺伝子oma-1(遺伝子ID WBGene00003864)は胚発生の2細胞期にほぼ独占的に発現するはずであり、遺伝子skn-1(遺伝子ID WBGene00004804)は発生にわたってかなり一貫した発現レベルを示すと予想されます。これらの遺伝子の発現を発生段階にわたってプロットする(マニュアルより)。applyををクリック。
視覚化された。

興味深いことに、oma-1の発現は、実は成虫(adults)で最も高い。これは、成虫の中には、2細胞期の未受精胚が大量に含まれているためと考えられる(マニュアルより)。
異なるグループの遺伝子が、ワームのライフサイクルの中でどのように発現レベルが変化するか調べる。そのために、チュートリアルでは3つのクラスタリング手法で調べる。
1,K-Medoids clustering
K-Medoidsクラスタリングは距離ベースのクラスタリング手法で、アルゴリズムはすべての遺伝子をK個のクラスタに分け、各クラスタは1つの遺伝子を中心としたものにしようと試みる。

このK-Medoidsクラスタリングでは、期待するクラスタ数(K)と、遺伝子間の発現の類似性を測定するための距離メトリックという2つの主要なパラメータを受け取る(マニュアルより)。
生物学的に意味のあるクラスターがデータ中にいくつ存在するのかがわからないため、期待するクラスターの数を正確に指定することは難しい。さらに、この数は、解析をどの程度細かくするかによって変わる。データセットに適した数のクラスターを提案するGap Statistic法を用いて、クラスター数のベターな選択肢を推定する。
パラメータ n_clusters の値を ”gap”にし、クラスター数を決定するためにGap Statisticsを使用したいことを示す。次に、距離メトリックを設定する。異なる距離メトリックは、特定のタイプのデータに対して、より効果的であったり、そうでなかったりす。RNAlysisは、RNAlysisのユーザーガイドに記載されているように、多くの距離メトリックを提供している。ここでは、時系列遺伝子発現データ用に特別に開発され、RNAlysisに実装されたYR1というあまり知られていない距離メトリックを使用する。Son and Baek 2007で詳しく紹介されている(マニュアルより)。

applyをクリック。計算が終わるまでしばらく時間がかかる(数分かかった)。
計算が終わると図が出力される。左上と左下はクラスタリング結果で、右下の図がGap Statistic アルゴリズムの結果となる。右下の図の左のグラフは、n_clusterの各値について、クラスタ内分散の自然対数(ln)を示している。この値は、クラスタが増えるほど下がることが予想される(クラスタが増えるほど、各クラスタに含まれる遺伝子は少なくなり、したがって各クラスタ内の遺伝子は互いに似てくる)。したがって、計算したクラスタリングソリューションの実際の分散値(青い線)と、同じ分布から引いたランダムデータのクラスタリングソリューションの分散値(オレンジ線)の両方を示している。右のグラフでは、観察された分散と期待された分散の比の自然対数を見ることができる- これがGap Statisticである。グラフの中の局所的な「ピーク」、つまり、隣のクラスタよりも大きなGap Statisticを持つn_clusterの値を探している。RNAlysisは、この基準に適合するn_clusterの最小値を自動的に選ぶが、その結果に基づいて他の良い値も提案する(マニュアルより)。
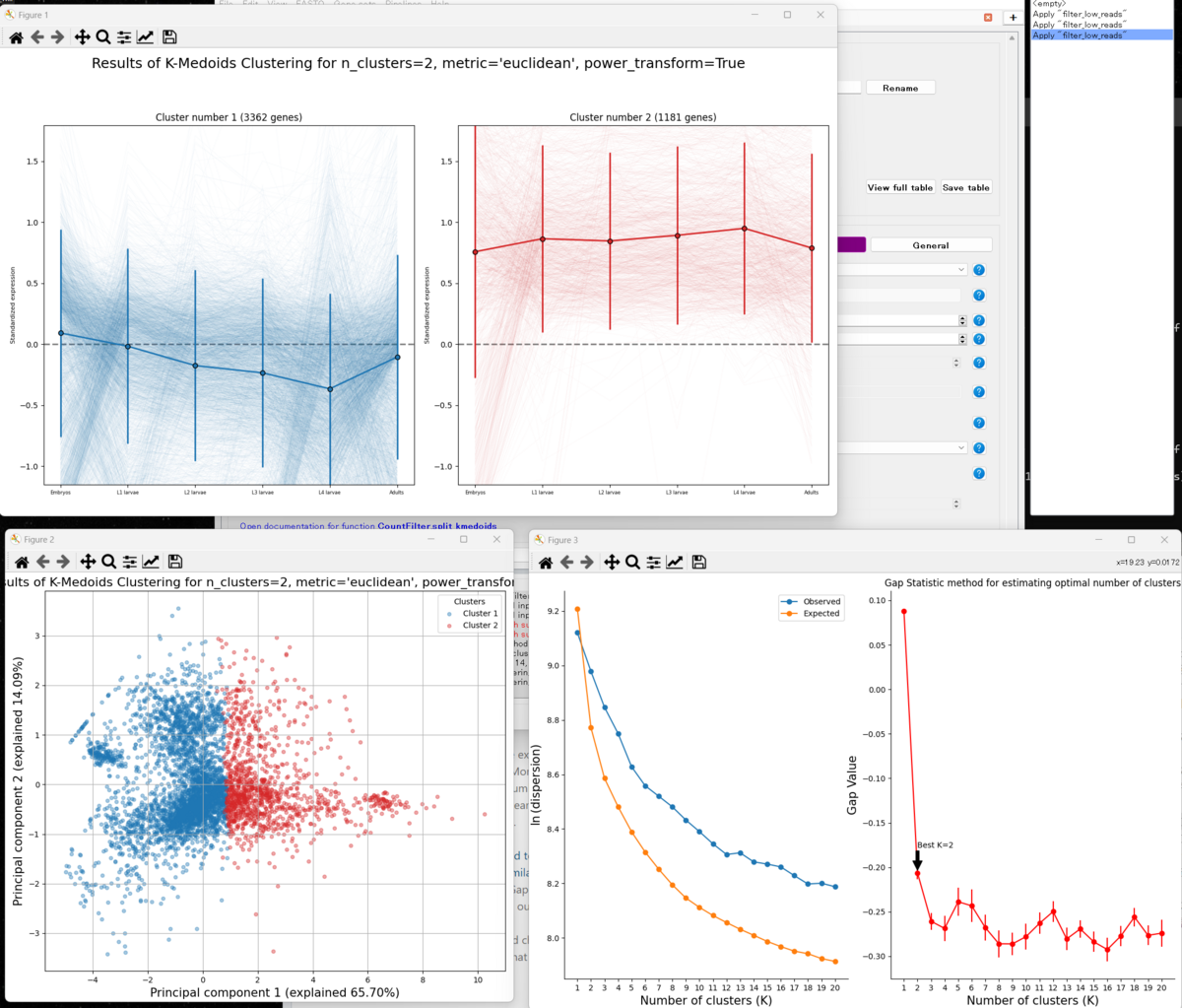
右下の図ではbestに2が提案されており(黒矢印)、K-Medoidsクラスタリングはクラスタ数2で実行された。その結果が左上と左下のグラフになる。
左上のグラフは2つのクラスタにおける遺伝子発現の分布を示している。絶対的な発現量(高発現/低発現遺伝子)ではなく、相対的な発現パターンによって異なる遺伝子をグループ化することに関心があるので、発現値は正規化され、べき乗変換されていることに注意する。左下の図では、遺伝子発現データの主成分分析投影を生成し、各クラスタ内の遺伝子を異なる色でマークする(ここでは青と赤のみ)。どこまでの主成分が遺伝子発現の分散の大部分を説明するか調べることができる。(マニュアルより)
計算後にはウィンドウが出現して、残す出力クラスタを選択するよう促される。また、残すクラスタに新しい名前を付けるオプションが表示される。

残すことを選択したクラスターごとに、そのクラスターに属する遺伝子だけを含むオリジナルのカウントマトリックスの新しいコピーが作成される。ここではクラスタ2つからタブが2個作成された。

今回はこれらのタブは閉じる。
2、HDBSCANによる密度ベースクラスタリング
HDBSCANは、クラスタリングアルゴリズムの異なるカテゴリーである密度ベースのクラスタリングに属します。HDBSCANはHierarchical Density-Based Spatial Clustering of Applications with Noiseの略です(出版物)。HDBSCANは、より伝統的なクラスタリング手法に対して複数の利点を提供する(マニュアルより)。
- HSBSCANはデータに関する仮定が比較的少なく、データにはノイズが含まれていると仮定し、また我々が発見したいと考える実際のクラスタも含まれていると仮定する。
- 他の多くのクラスタリング手法とは異なり、HDBSCANはすべての遺伝子をクラスタに属するように「強制」するわけではない。その代わりに、遺伝子を外れ値として分類し、最終的なクラスタリングソリューションから除外する。
- HDBSCANを使用する場合、データ中のクラスタ数を推測する必要はない。HDBSCANの主な調整パラメータは最小クラスタサイズ(min_cluster_size)で、これはデータから見つかると予想される最小の「興味深い」クラスタサイズを決定する。
HDBSCANを実行するには、min_cluster_sizeの値を選択する必要がある。min_cluster_sizeの値を小さくすると、より多くの小さなクラスターが返され、遺伝子発現データのより細かなパターンを明らかにする。min_cluster_sizeの値が高いほど、大きなクラスターの数が少なくなり、データ中の最も明白で重要なパターンを明らかにする。
ここでは75とする。距離法はYR1を選択。

applyをクリック。計算は10秒以内に終わった。
HDBSCANは1の方法よりもはるかに多くのクラスタを生成しており、クラスタ毎にかなり一貫性があることがわかる。

1と同様に、各クラスタのカウントを別のタブで保存するかどうか選ぶことができる。
3、複合的なアプローチ - CLICOMによるアンサンブルベースのクラスタリング
emsembleベースのクラスタリングアルゴリズム。CLICOM(出版物)は、異なるクラスタリングパラメータを持つ複数のクラスタリング結果を組み込み、コンセンサスクラスタリングソリューションを作成する。CLICOMは、従来のクラスタリング手法に比べて複数の利点を備えている(マニュアルより):
- アンサンブルクラスタリングのアプローチでは、複数のクラスタリングアルゴリズムと複数のチューニングパラメータの結果を組み合わせることができ、個々のクラスタリング手法の弱点を補う可能性があり、多くのクラスタリングソリューションに頑健に現れるパターンのみを考慮することができる。
- CLICOMを使用する場合、データ中の最終的なクラスタ数を推測する必要はない。HDBSCANの主なチューニングパラメーターはエビデンス閾値(evidence_threshold)。
RNAlysisはCLICOMの修正版実装を提供している。このアルゴリズムの修正版は、HDBSCANアルゴリズムと同様に、遺伝子を外れ値として分類し、最終的なクラスタリングソリューションから除外することができる。さらに、このアルゴリズムの修正版は、データ中の生物学的/技術的複製物の各バッチを別々にクラスタリングすることができる。これは、クラスタリング結果に対するバッチ効果の影響を低減し、クラスタリング結果の精度と堅牢性を向上させる。(マニュアルより)。
CLICOMアルゴリズムを選択する。

ウィンドウが表示される。このウィンドウの左半分では、CLICOMアルゴリズムのチューニング・パラメータを設定する。チュートリアルでは、evidence thresholdを0.5に設定し、最小クラスタサイズを75に設定している。

チュートリアルのデータには反復がないので、replicates_groupingパラメータを調整する必要はない。
- evidence_threshold:2つの遺伝子が一緒にクラスタリングされていることについて、最終的な解決策で一緒に表示されるために、いくつのクラスタリングソリューション(0-1の間の割合)が同意しなければならないか?証拠閾値が低いほど、大きなクラスターが少なくなり、外れ値として分類される特徴が少なくなる。
- cluster_unclustered_features: Trueに設定すると、CLICOMはすべての遺伝子を発見されたクラスターに属するように強制する(アルゴリズムのオリジナル実装と同様)。そうでない場合、遺伝子はノイズとして分類され、クラスタ化されないままにできる。
-
min_cluster_size: 意味のあるクラスターの最小サイズを決定する。これより小さいクラスターはノイズとして分類され、最終結果からフィルタリングされるか、他のクラスターにマージされる(cluster_unclustered_featuresの値による)。
-
replicates_grouping: サンプルを技術的/生物学的バッチにグループ化することができる。このアルゴリズムは、各バッチのサンプルを個別にクラスタリングし、CLICOMアルゴリズムを使用して、すべての個別のクラスタリング結果からアンサンブルクラスタリング結果を見つける。
ウィンドウの右半分では、CLICOMの実行に異なるクラスタリングパラメータを持つクラスタリング・セットアップを追加する。RNAlysisで利用可能なクラスタリング・アルゴリズムのいずれでもよく、同じアルゴリズムを使った複数のクラスタリング・セットアップを含め、複数追加できる。セットアップを追加するには、ドロップダウンメニューからアルゴリズムを選び、パラメータを設定し、「Add Setup」ボタンをクリックする。

追加したセットアップは右側のウィンドウに表示される。

アンサンブルアプローチのため、チュートリアルではK-Medoidsクラスタリング(7クラスタ、Spearmanメトリック)、階層的クラスタリング(8クラスタ、ユークリッドメトリック、Ward linkage)、HDBSCANクラスタリング(Jackknife distanceメトリック、最小クラスタサイズ150)を追加している。

最後にCLICOM開始ボタンをクリックする。
10秒ほどで図が出力された。

計算終了時に表示されるパネルから、タブに保存したいクラスタを指定できる。ここでは3つとも残した。
興味のあるクラスターを抽出できたら、エンリッチメント解析によって生物学的意義に関する知見を得る。Gene setsメニューのEnrichment Analysis選択。

基本的なエンリッチメント解析では、まずエンリッチメントセット(興味のある遺伝子セット、例えば「L1から成体へのダウン」)とバックグラウンドセット(エンリッチメント結果を比較する参照遺伝子、例えばフィルターした元のテーブルの遺伝子)を選択する。Enrichmentウィンドウの2つのドロップダウンメニューから選ぶ。

次にデータセットを選択。ここではGOを選ぶ。

データセットを選択するとウィンドウの下側が拡大され、すべてのパラメータが表示される(画像は展開後)。
統計検定にhypergeometric、生物にCaenorhabditis elegans(遺伝子発現データと一致させる)、Gene ID TypeにWormBase(元のデータテーブルの遺伝子ID Typeと一致させる7)を選ぶ。

画像は生物を選んでいるところ。初期で画像の生物をサポートしている。
Gene ID Typeはより多くのタイプをサポートしている。WormBaseを選択。

残りの設定はデフォルトのままで進める。runボタンをクリック。
計算が終わるまで1分ほどかかった。結果は複数のフォーマットで表示される。
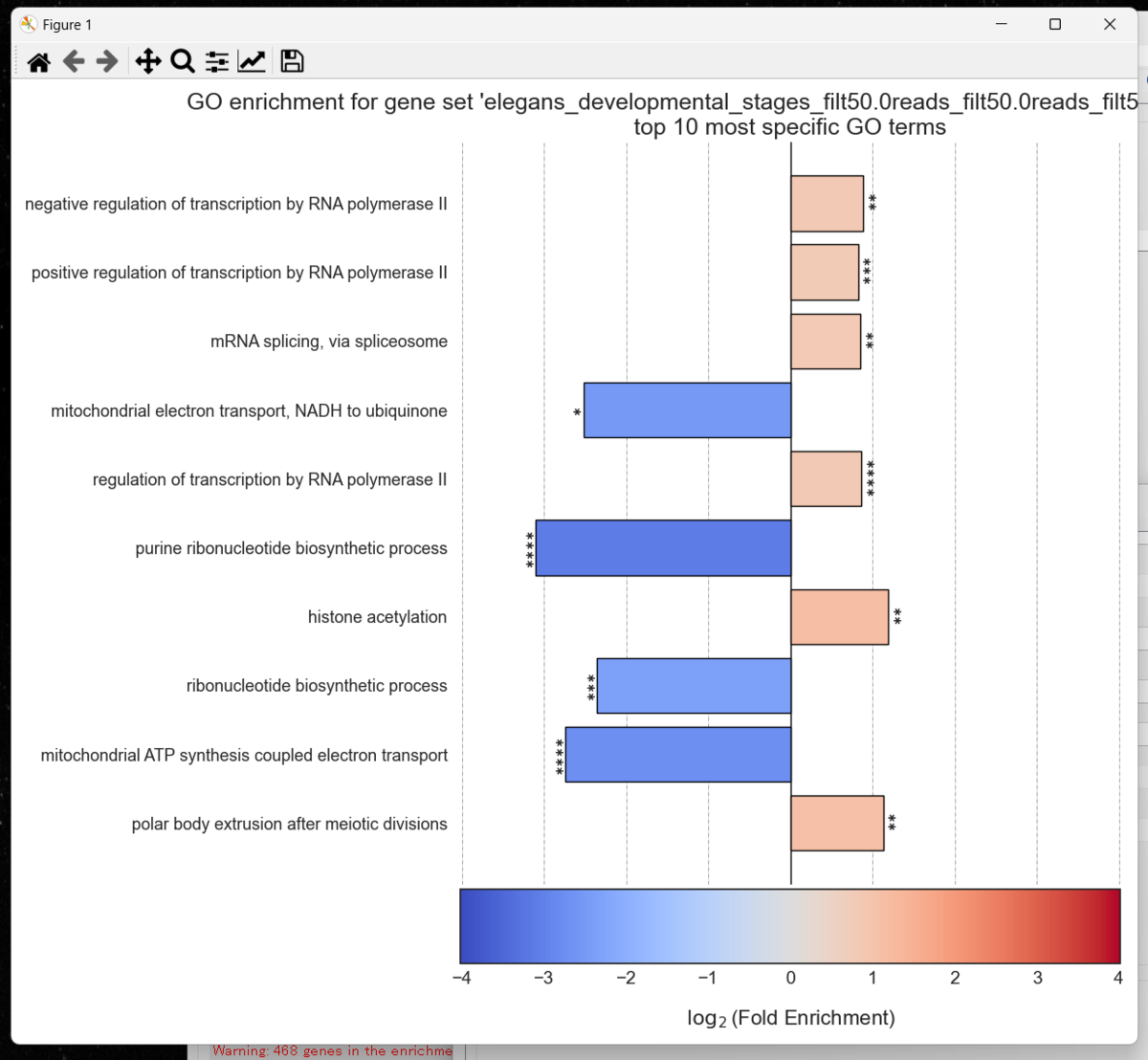
表形式で、統計的に有意なGO term(return_nonsignificantパラメータをTrueに設定した場合は、テストしたすべてのGO term)表示される。

表は特異性でソートされている。他にも、クラスタ内の遺伝子数、GO termに一致する遺伝子数、GO termに一致する期待遺伝子数、log2 fold change、p値、および調整p値が含まれている。
2つ目の出力はオントロジーグラフ。3つの各GOカテゴリにおいて統計的に有意なGO termと、オントロジーグラフにおけるその祖先が描かれている。グラフ内のGO termの階層は、公式のGene Ontologyの階層と一致している。

グラフ上のtermの色はlog2倍変化を示し、ツリーでより特異的なGO termは下部にある。パネルは上の虫眼鏡ボタンで拡大でき(ドラッグで囲む)、ホームボタンで元のサイズに戻せる。
3つ目の出力は最も統計的に有意な10個のGO termのlog2倍率変化と有意性(色で表す)を示すバープロット。
チュートリルの2つ目の解析では、GEOから公開されている3つの線虫のRNA-seq実験を使用している。この実験では、遺伝子発現に対する特定のストレス条件の影響が調べられている。目的は、異なるストレス条件間の類似性を調べ、特定の疑問、すなわち、エピジェネティックな遺伝子や経路がストレスにさらされるとどのような影響を受けるのかに答えることにある。2つ目のチュートリアルでは、GEOで公開されている生のFASTQファイルから直接、各データセットを前処理して定量を行う(チュートリアルより)。
他にも様々な機能があります。簡単に見ていきます。
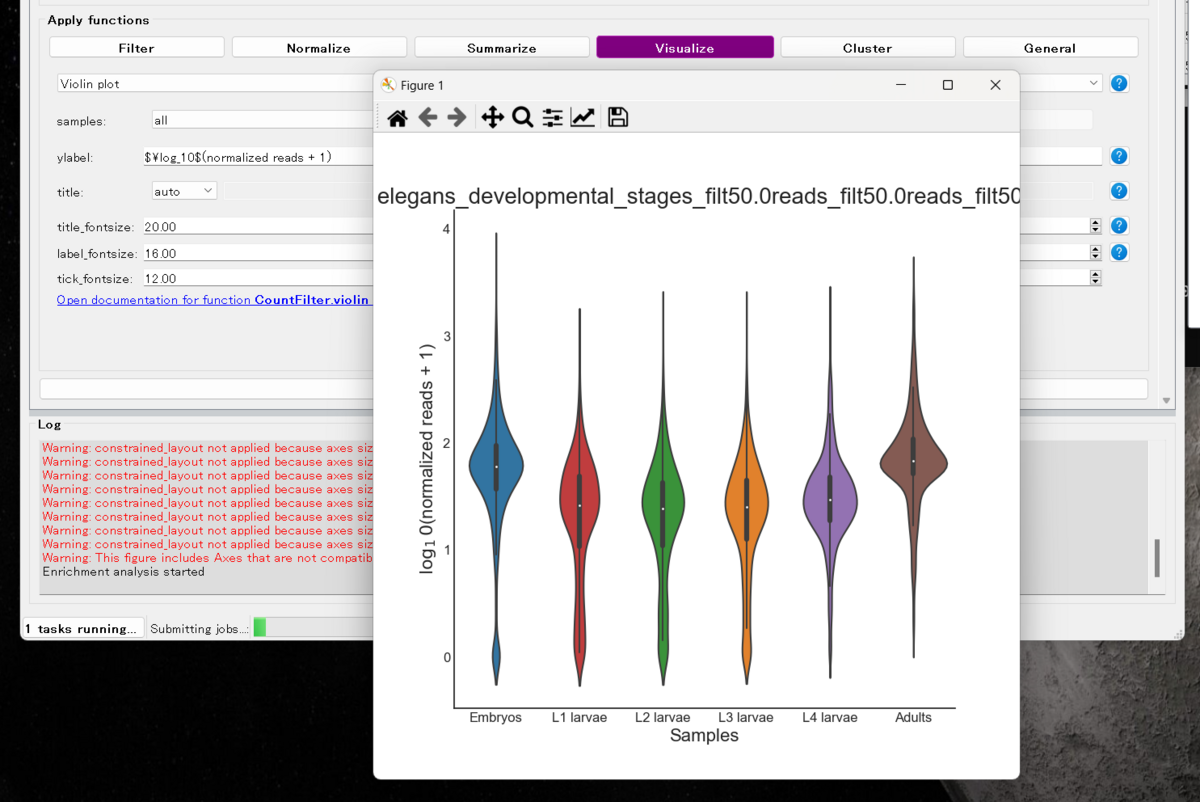
Visualize => violin plot
Visualize => Scatter plot

比較するペアを指定する。
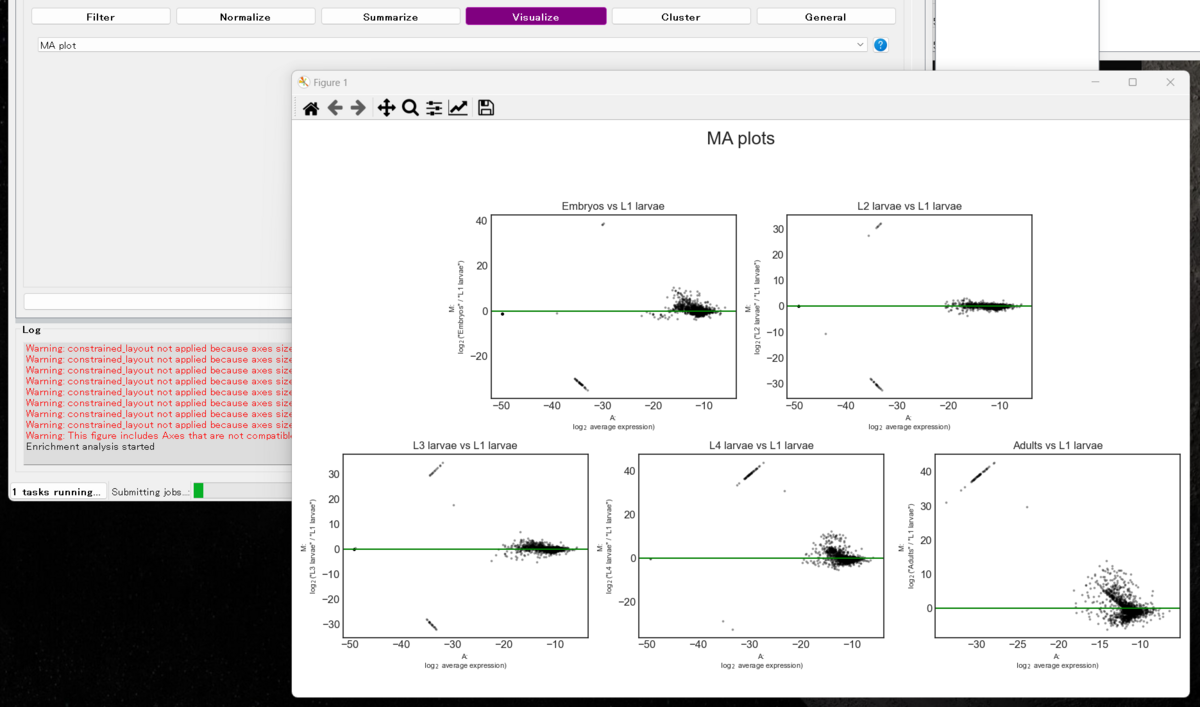
Visualize => MA plot
Visualize => Hierachical clustergram plot
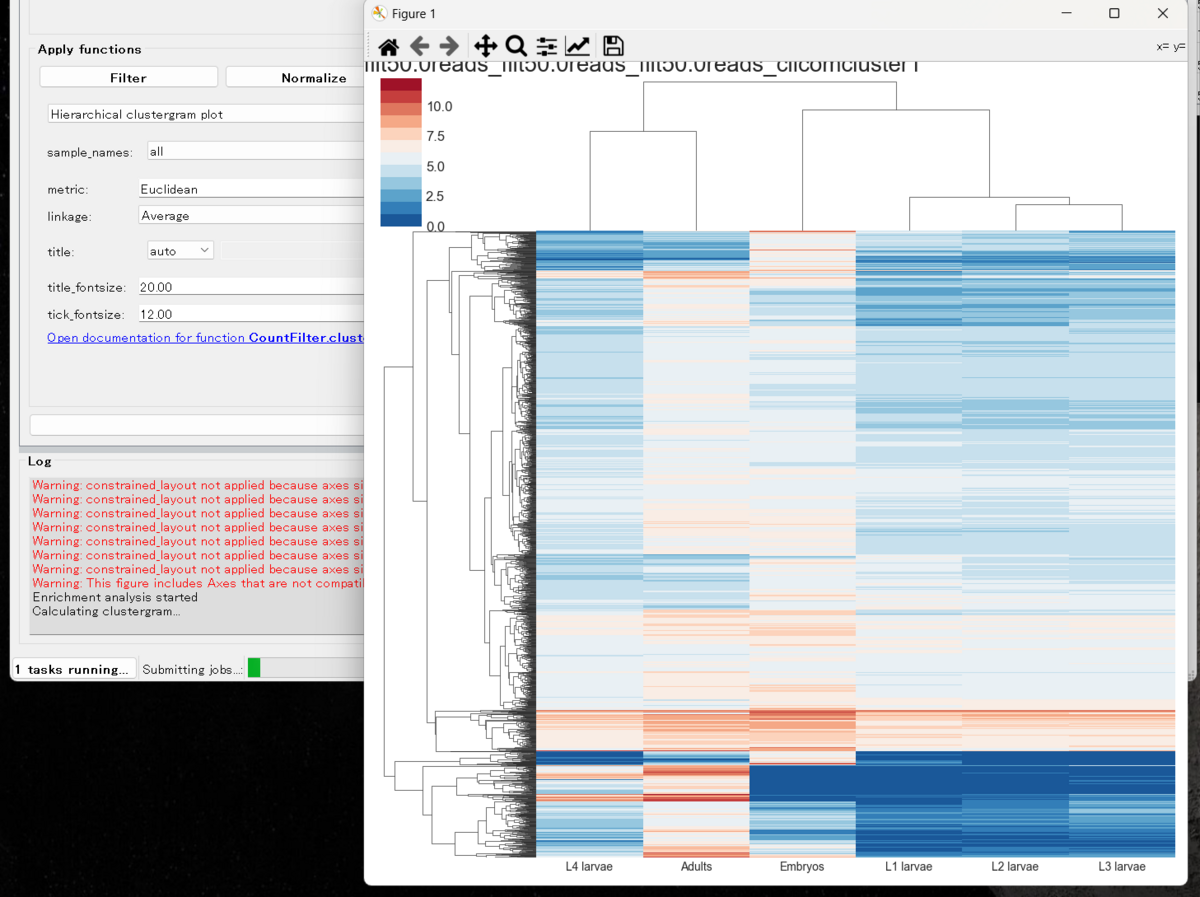
ユークリッド距離、average linkage
Visualize =>Enhanced box plot
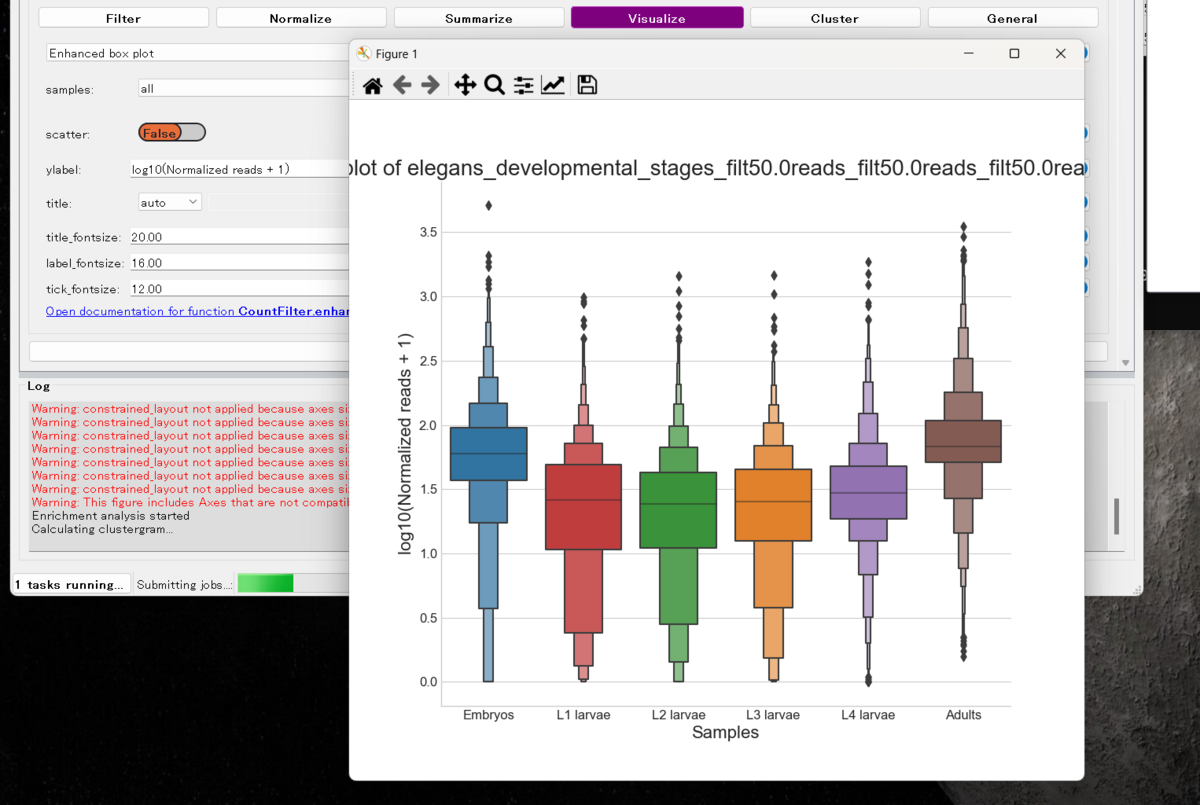
log10変換
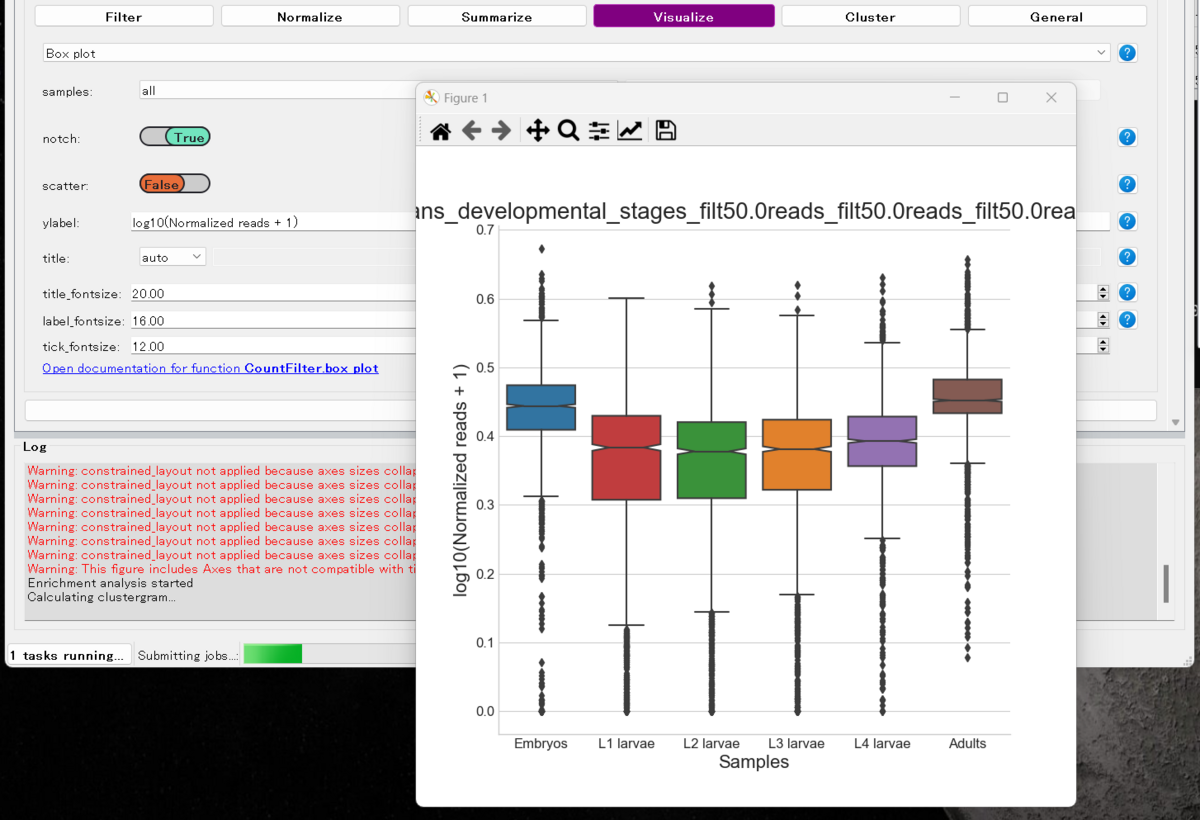
Visualize => Box plot
引用
RNAlysis: analyze your RNA sequencing data without writing a single line of code
Guy Teichman, Dror Cohen, Or Ganon, Netta Dunsky, Shachar Shani, Hila Gingold, Oded Rechavi
bioRxiv, Posted November 25, 2022
関連