ヒトの健康や病気の形成に腸内細菌叢が果たす複雑な役割は、培養に依存しない分子ベースのハイスループットシーケンシング技術が利用できるようになったこともあって、近年、精力的に調査・研究が行われている。ヒトのすべての宿主は、平均500~1000種の異なる細菌と共存していると推定されており[ref.1,2,3]、マイクロバイオームが宿主のライフスタイルや食生活[ref.4,5]、さらには肥満、2型糖尿病[ref.6]、ガン[ref.7]などの多くの疾患と関連していることが研究によって明らかになってきている。新しいシーケンス技術は、マイクロバイオーム研究のためのより多くのデータと能力をもたらすだけでなく、データ分析と解釈のための新たな課題ももたらす。マイクロバイオームデータ解析のためのツールや手法が改善されれば、多様な環境における微生物の役割を理解する能力が向上し、特に微生物同士や宿主である人間との相互作用を理解することができる。
現在のマイクロバイオーム解析は一般的に,上流のコミュニティプロファイリング(例:各サンプルに含まれるすべての微生物の存在量)と下流のハイレベル解析(例:アルファ/ベータ多様性解析,存在量の差の解析)という2つの重要な要素で構成されている[ref.8]。近年、進化したデータ解析、可視化、機械学習の手法が徐々に適用され、これら2つの要素をカバーするマイクロバイオームデータ解析用の多くのソフトウェアツールやウェブサーバが開発されている[ref.9,10,11,12,13]。しかし,新しい技術やシーケンス技術により,マイクロバイオームデータの解析と解釈に新しい手法を適用する科学研究者の学習曲線は急峻になっている[ref.14]。さらに,既存のツールは,ほとんどが解析の一面に特化しており,また,1種類のマイクロバイオームデータの解析に限定されている.例えば,16S rRNA データを解析するためのツールやワークフローは数多く存在するが,RNA ベースのメタトランスクリプトミクスの解析ニーズに包括的に対応するためのツールやパイプラインは存在しない。
論文表1は、マイクロバイオームデータの解析ニーズに対する、これらのツールの機能をまとめたものである。16S rRNAのようなマーカー遺伝子ベースのデータについては,QIIME II [ref.15]やMothur [ref.16]がユーザーインターフェースと豊富な解析・可視化ツールを提供しているが,メタゲノムやメタトランスクリプトームのデータには対応していない。Vegan [ref.17]は、メタゲノムデータの可視化のための様々な機能を提供しているが、ユーザーインターフェースや、宿主や微生物のリードアラインメント、差分発現などのツールがない。BioBakery [ref.18]は、微生物群集のほとんどのメタゲノム解析ニーズに対応した包括的なツール群を提供しているが、種の同定には少数のマーカーセットに依存しており、宿主や微生物の発現には対応していない。Microbiome helper [ref.19]は、マイクロバイオームおよびメタゲノム解析ツールの相互運用を容易にするための複数言語によるスクリプト集であるが、インタラクティブな可視化やグラフィカル・ユーザー・インターフェースは提供していない。Rのmicrobiomeパッケージ[ref.20]は,メタゲノムデータ解析の様々なタスクに対応したコマンドラインワークフローを提供している。Phyloseq [ref.21]は,アノテーション,可視化,多様性解析のためのツールを備えたShinyインターフェースを持っているが,アバンダンス解析は提供しておらず,開発者による積極的なメンテナンスも終了している。Metavizr [ref.22]は、メタゲノムの視覚化のためのインターフェースと機能を提供している。これらの手法はいずれも包括的ではなく、複数のタイプの16S rRNA、メタゲノム、メタトランスクリプトームデータのニーズに特化していない。そのため、マイクロバイオームデータの解析と解釈のための完全なワークフローを含む既存のツールキットは存在しない(グラフィカルユーザーインターフェースの有無は問わない)。
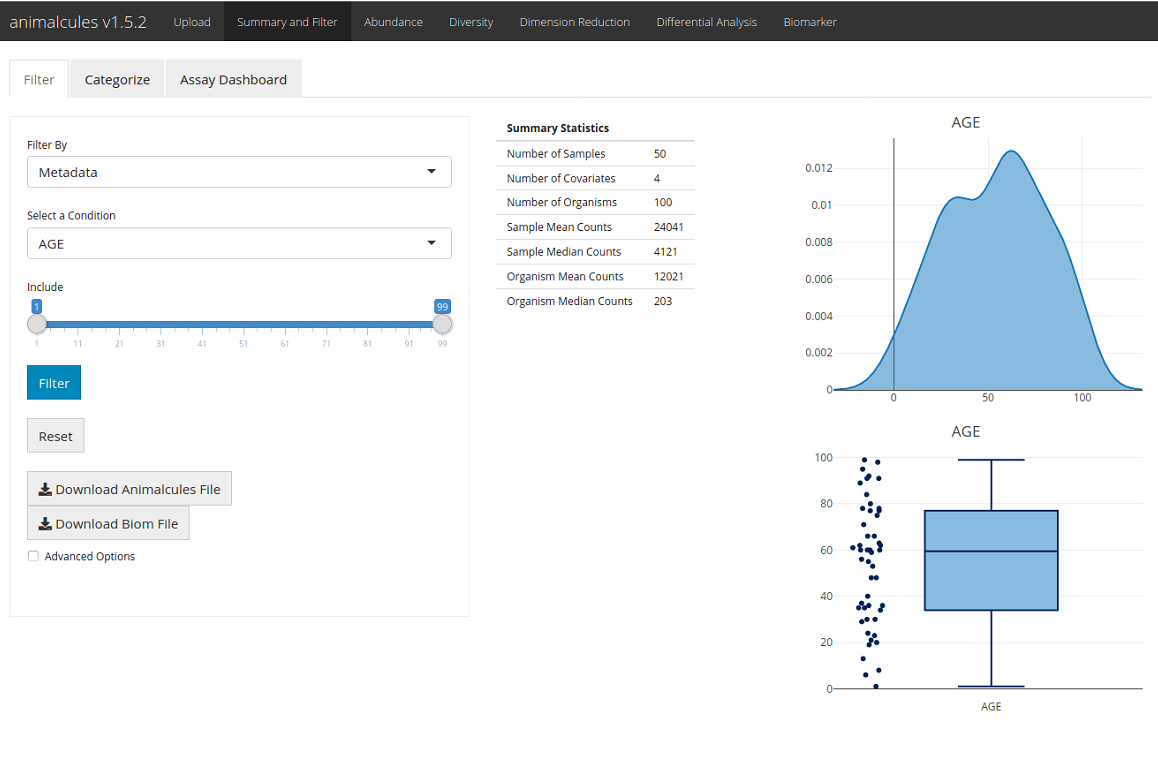
animalculesは,種のカウントテーブル,organizational taxonomic unit(OTU)やアンプリコン配列バリアント(ASV)カウントテーブル,Biological Observation Matrix(BIOM)形式など,複数の形式のマイクロバイオームプロファイルのインポートをサポートしている[ref.23].これらのフォーマットは、16S rRNA、メタゲノミクス、メタトランスクリプトミックデータなど、一般的なマイクロバイオームデータソースや分析ツールから生成することができる。データをアップロードすると、animalculesは便利なデータサマリーとフィルタリング機能を提供し、ユーザーはサンプルのメタデータ、微生物の有病率、または相対的な存在感を用いてデータセットを表示およびフィルタリングすることができる。このようにデータをフィルタリングすることで、前処理や下流の解析作業にかかる時間を大幅に短縮することができる。相対存在量の棒グラフや3次元リダクションプロット(PCA/PCoA/tSNE/UMAP)などのデータの可視化については、animalculesは、ユーザーが各データポイントのサンプル/微生物情報を確認し、必要に応じて図のフォーマットを調整できるインタラクティブな操作をサポートしており、サンプルサイズや微生物の数が多い場合に要素やデータパターンを認識するのに役立つ。animalculesは、一般的な多様性分析、ディファレンシャル・アバンダンス分析、次元削減に加え、ロジスティック回帰またはランダム・フォレスト・モデルを学習し、クロスバリデートされたバイオマーカーの性能評価を行うことで、バイオマーカーの同定をサポートする。animalculesは、R/Shinyによるグラフィカル・ユーザー・インターフェース(GUI)を提供しており、プログラミングの知識がないユーザーでも使用することができる。経験豊富なプログラマーは、コマンドラインベースのRパッケージを選択するか、またはその両方を組み合わせることができる。
animalculesのすべてのデータ処理タスクおよび関数は、MultiAssayExperiment(MAE)データ構造に基づいており、それを用いて動作する。MAEクラスは、複数のアッセイにまたがるサンプルの連結をサポートする効率的なデータ検索および操作方法を備えた、マルチオミクス実験のための標準的なデータ構造である。MAEオブジェクトは、colData(被験者またはセルラインレベルのメタデータを含む)、ExperimentList(1つまたは複数のアッセイのデータを格納)、sampleMap(実験とサンプルを関連付ける)の3つの主要コンポーネントを備えている。animalculesでは、3つのテーブル(サンプルメタデータテーブル、微生物数テーブル、タクソノミーテーブル)とそれらの間のマッピング関係がMAEクラスに格納されている。これにより、アッセイや被験者の正しい配置が保証され、サンプルやフィーチャーの調整されたサブセットが提供される。さらに、多くのBioconductorパッケージで適用されているSummarizedExperimentクラスからMAEオブジェクトへの変換やMAEオブジェクトからの変換が容易で、他のツールとのスムーズな連携が可能である。MAEクラスをマイクロバイオーム研究分野に適用することの重要な利点の1つは、多くのマルチオミクスレイヤーのデータをサポートする拡張可能な設計である。animalculesは、MAEオブジェクトを統合した初めてのマイクロバイオーム解析用ソフトウェアツールであり、MAEのユニークな特性を活かして、微生物データ、宿主のトランスクリプトミクス、メタボロミクス、さらには分類学上の情報を同じオブジェクト内に格納することができる(現在、animalculesはマイクロバイオーム解析のみをサポートしているが、MAE構造により、これらのデータタイプに対応できる将来の開発が可能になる。さらに、MAEは、これらのデータタイプを管理する他のツール(例:ホストトランスクリプトミクス)との統合を可能にする。) MAEオブジェクトは、様々なアッセイの処理済みバージョン(例えば、次元削減されたデータ)を保存することができ、下流での効率的な操作や分析を可能にする。このアプローチは、必要とされる様々なマルチオミクスデータセットを効率的に統合することで、標準的なマイクロバイオーム解析とデータ共有を促進する。
animalculesはR ≥ 4.0.0を必要とし、GitHub(https://github.com/compbiomed/animalcules)から自由にダウンロードでき、Bioconductor(https://www.bioconductor.org/packages/release/bioc/html/animalcules.html)からインストールすることができる。
https://compbiomed.github.io/animalcules-docs/
commandline interface
オーサーから提供されているdocker imageを使用してテストした。
R Shinyインターフェース
docker pull bucbm/animalcules:latest
docker run -d -p 8787:8787 -e PASSWORD=animalcules bucbm/animalcules:latest
http://localhost:8787/ にアクセスする。
username: rstudio
password: animalcules
Rstudioにログイン後、animalculesをロードする。
library(animalcules)
run_animalcules()
立ち上がった。
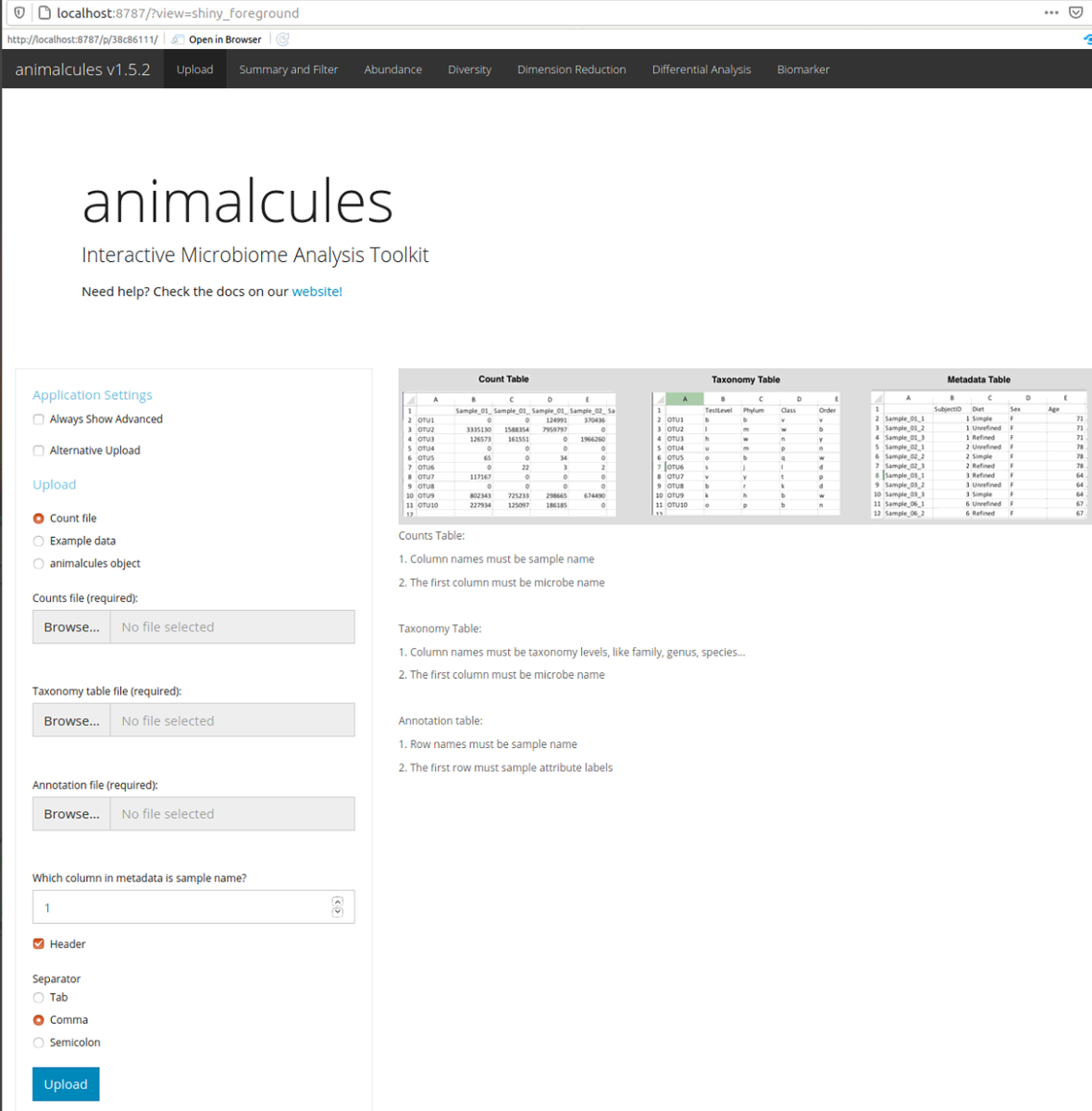
データのアップロード方法は8種類ある。
- Count File(主に新しいデータセットをアップロードする際に使用)。
- exampleデータ (アニマルキュールを調べるためのデータ)
- animalculesファイル(以前にアップロードしたデータセットを素早く再アップロードする)。
- phyloseqオブジェクト
- BIOMファイル
- taxonomy ID付きカウントファイル(taxonomy テーブルが利用できない場合のオプション)
- 病原体ファイル
- animalcules-IDファイル
Count Fileについては、16sおよびメタゲノミクスで生成されたデータの両方がサポートされている。必要なファイルは以下の3つになる。
1、Countsファイル:各行は種/OTU、各列はサンプル名。
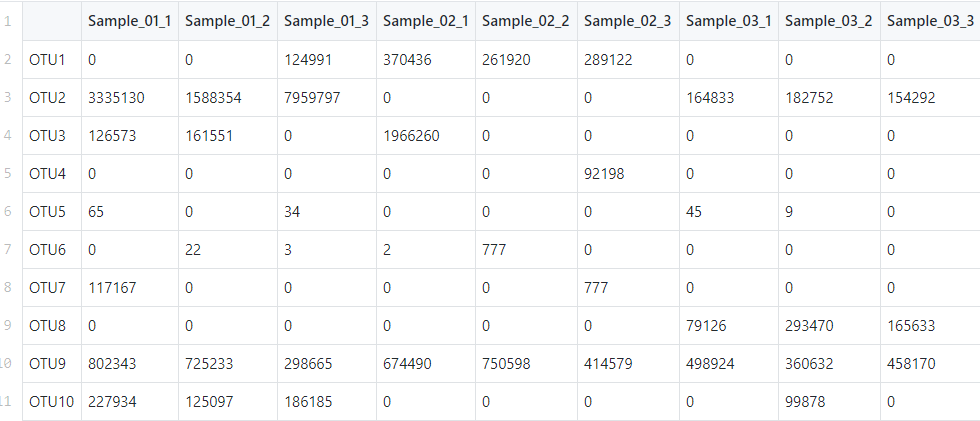
animalcules-docs/count_example.csv at master · compbiomed/animalcules-docs · GitHub
2、taxonomy ファイル:各行は種/OTU、各列はtaxonomy レベル。
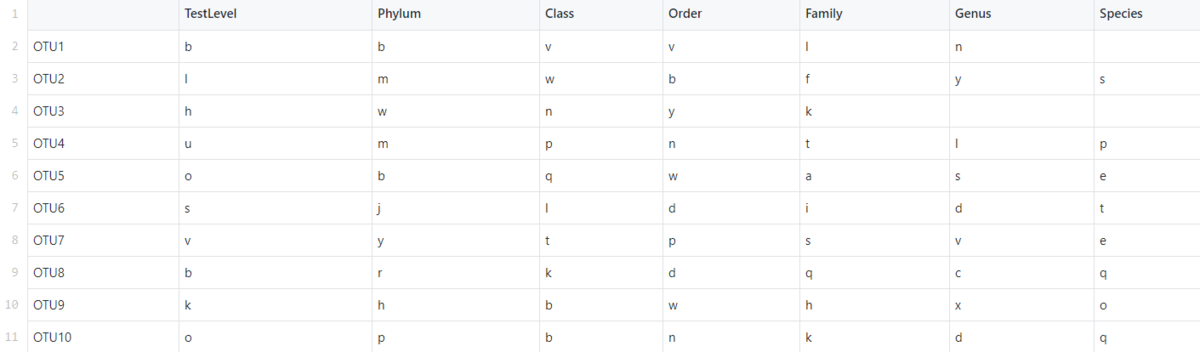
animalcules-docs/taxonomy_table.csv at master · compbiomed/animalcules-docs · GitHub
3、注釈ファイル:各行はサンプル名、各列は変数/特徴名。

animalcules-docs/annotation_example.csv at master · compbiomed/animalcules-docs · GitHub
3つのファイルのサンプル名、微生物名は一致している必要がある。また、3つのファイルの同じファイルタイプ(例:csv)は同じでなければならない。
用意が出来たらアップロードする。
タブ区切り、カンマ区切り(デフォルト設定)、セミコロン区切りのテキストファイルをアップロードできる。
ここではexampleデータを使う。

example datasetは3種類;シミュレーションデータセット(デフォルトでロード済み)、結核データセット、喘息データセット(metatranscriptomics)、用意されている。ここではSimulated datasetを選択した。
分析結果は自動では出力されない。データのアップロード後、上のボタンから各タブに切り替えて調べていく。
簡単に視覚化結果だけ見ていきます。
Summary and Filterタブ
Abundanceタブ
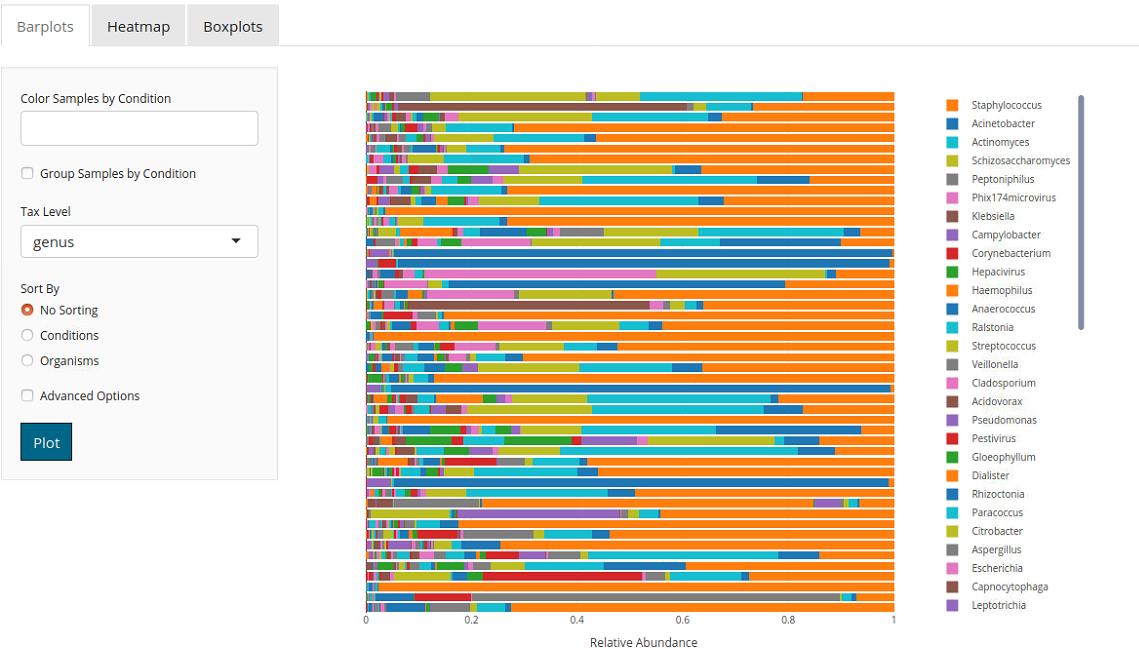
ある分類レベルの微生物の相対的な存在量を視覚化
Barplot

横向きのバー1つで1サンプルに相当。上から順に数十サンプル表示されている。
phylumからgenusレベルに切り替えた。
Heatmap

Boxplot
複数の分類レベルにおける1つまたは複数の生物の存在量をカテゴリー属性間で比較

画像ではgenusレベルで1つの分類群のみ比較している。複数追加して横に並べることが可能。
Diversityタブ
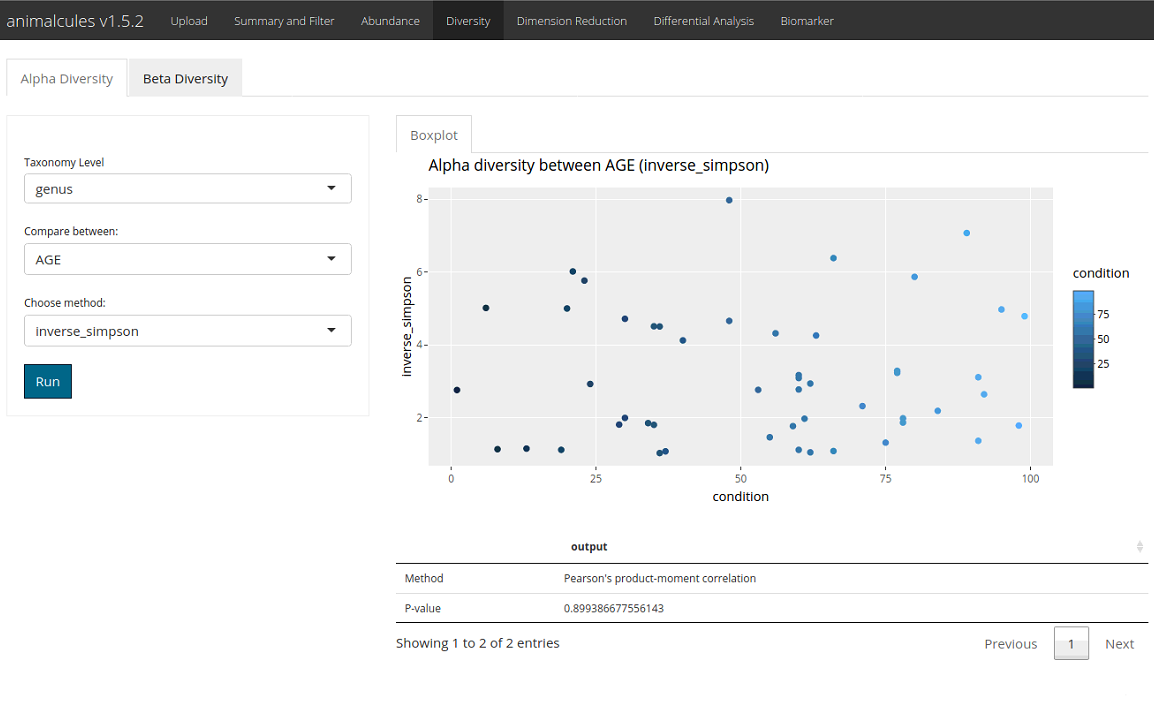
詳細オプションでは、Shannon、Inverse Simpson、Gini Simpsonなどの異なるアルファダイバーシティメトリクスを選択できる。
比較をAGEからGROUPに変えた。
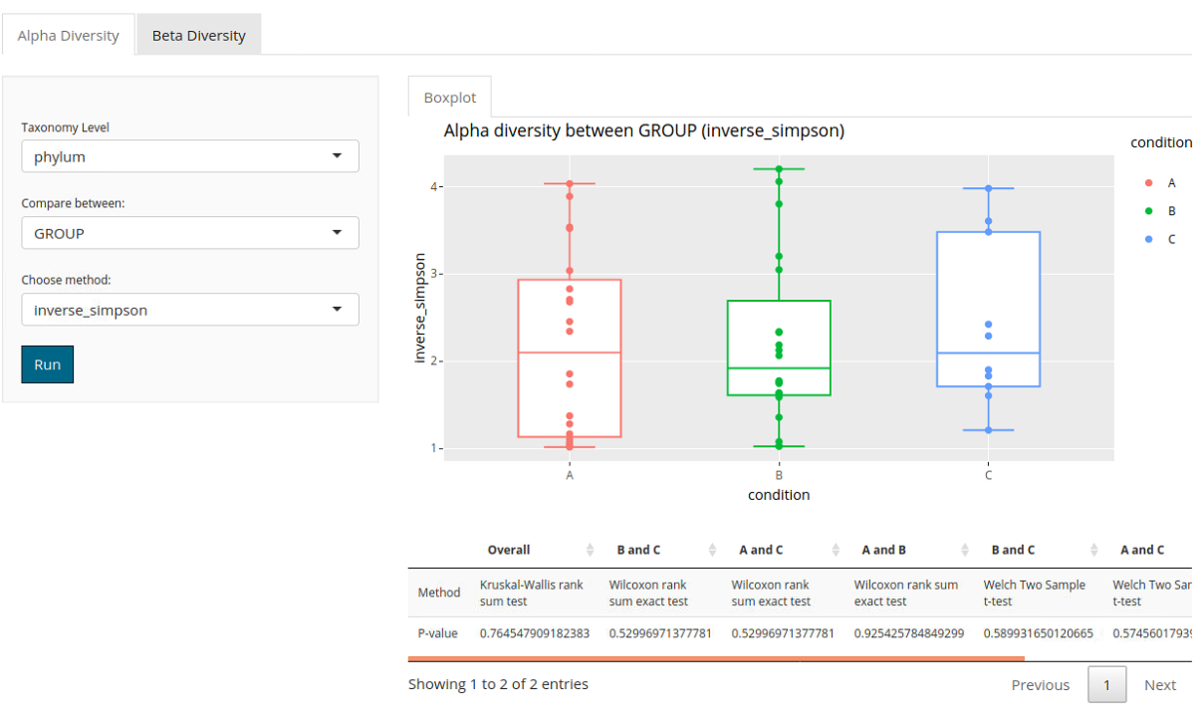
検定は、選択された対象条件におけるレベル間で実行される。T-検定、Kruskal-Wallis順位和検定、Wilcoxon順位和検定など、複数の統計検定法が自動的に適用される。検定のp値が結果の表に表示される。
Dimension Reductionタブ
2次元および3次元の主成分分析を行う
PCA
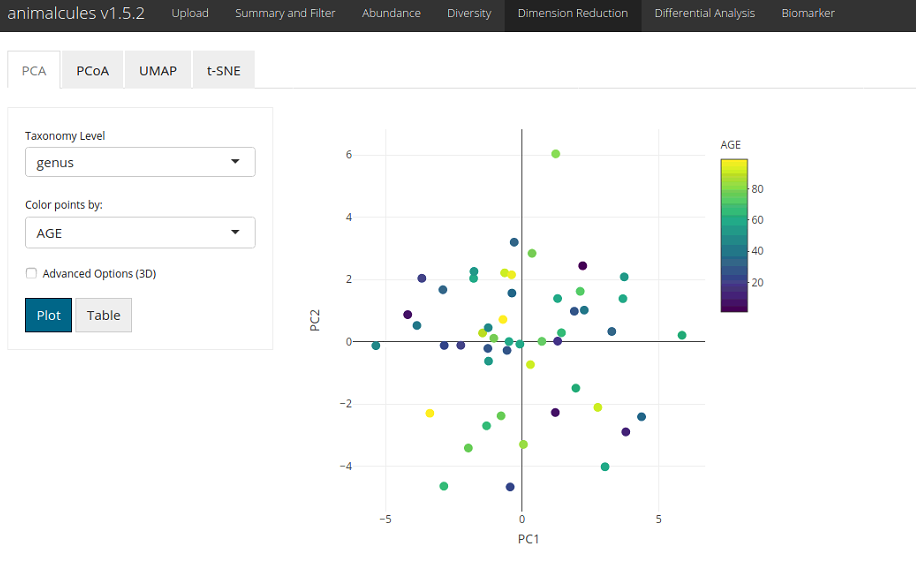
UMAP

t-SNE

第3主成分を追加して3Dプロット。

Differential Analysisタブ
Differential Abundance Analysisを特定の変数に対して行い、キーとなる微生物を特定する。
DESeq2
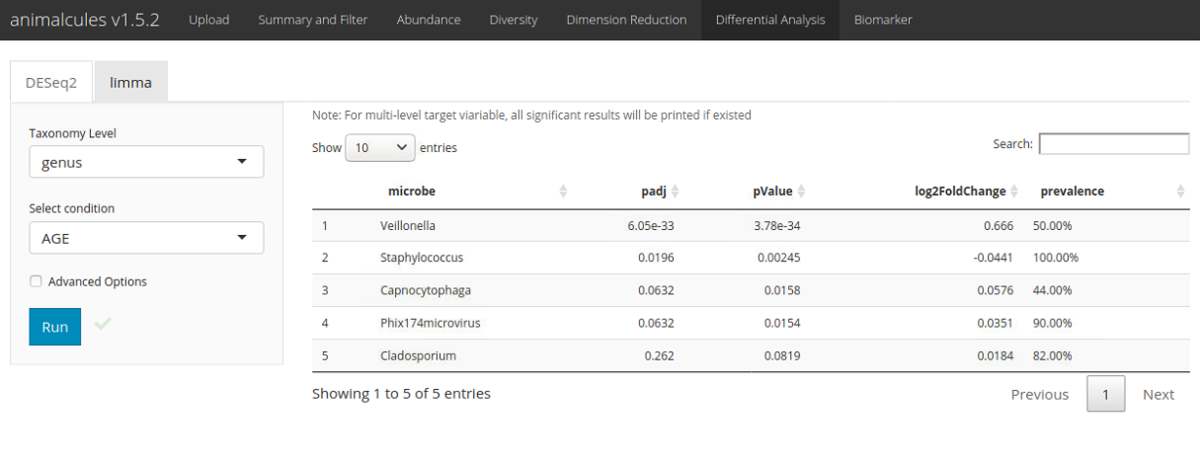
Advanced optionで線形モデルに共変量を追加したり、最小カウントカットオフを追加したり(平均リード数がこのカットオフより小さいフィーチャーをフィルタリング)、調整済みp値カットオフを追加したりできる。

limma

Biomarkerタブ
特定の二値分類問題に対するバイオマーカーを特定する
animalculesのユニークな機能の一つに、バイオマーカー(wiki)同定モジュールがある。ユーザーは、微生物のバイオマーカーを同定するために、ロジスティック回帰またはランダムフォレストのいずれかの分類モデルを選択できる。バイオマーカーの予測性能を評価するためのAUC値と平均クロスバリデーションROC曲線に加えて,各微生物のthe feature importance scoreが提供される。

importance plot

CV ROC plot

Biomarkerの詳細はDocumentを確認して下さい。
https://compbiomed.github.io/animalcules-docs/articles/rmd/biomarker.html
引用
animalcules: interactive microbiome analytics and visualization in R
Yue Zhao, Anthony Federico, Tyler Faits, Solaiappan Manimaran, Daniel Segrè, Stefano Monti & W. Evan Johnson
Microbiome volume 9, Article number: 76 (2021)
Animalcule
アニマルキュール(ラテン語のanimal+diminutive suffix -culumからなる「小さな動物」)とは、細菌や原生動物、非常に小さな動物などの微細な生物を指す古い言葉である。
https://en.wikipedia.org/wiki/Animalcule
関連