2021 2/17 tips追記
IGVをより便利に使う方法はないでしょうかという質問があったので、今日は自分が知っているIGVのtipsをいくつか紹介します。
IGVの公式動画チャンネルがあります。
統合TVでもIGVの基本的な使い方について分かりやすく説明されています
インストール
IGVのCUIへの導入はcondaで出来ます。ここでは高速なmambaを使います。
mamba install -c bioconda -y igv
> igv --help
Command line options:
Space delimited list of data files to load
--preferences, -o Path or URL to a preference property file
--batch. -b Path or url to a batch command file
--port, -p IGV command port number (defaults to 60151)
--genome, -g Genome ID (e.g hg19) or path or url to .genome or indexed fasta file
--dataServerURL, -d Path or url to a data server registry file
--genomeServerURL, -u Path or url to a genome server registry file
--indexFile, -i Index file or comma delimited list of index files corresponding to data files
--coverageFile, -c Coverage file or comma delimited list of coverage files corresponding to data files
--name, -n Name or comma-delimited list of names for tracks corresponding to data files
--header, -H http header to include with all requests for list of data files
--igvDirectory Path to the local igv directory. Defaults to <user home>/igv
--version Print the IGV version and exit
--help Print this output and exit
様々なオプションが利用できます。たとえば-l (--locus)オプションを使うと、指定の染色体ポジションを初期位置にして起動できます。
Tips
1、マウスホバーした時にポップアップウィンドウが出ないようにする。
上のメニューの吹き出しマークをクリックしてNever~を選択します。show Details on clickを選ぶと、クリックした時に大型のポップアップウィンドウが表示されます。

2、読み込んだデータの環境を保存する。
IGVに複数のデータを読み込んで細かく設定を調整したら、後になって再びその環境を再現するのが難しくなります。その時はxmlのセッションファイルをexportしておくと、次回はそれを読み込むだけで環境が完全に再現されます。
IGVで表示を色々チューニングしたとします。

File => Save Sessionを選択して、xmlファイルを保存します。

xmlファイルを保存したらIGVを終了します。
再びIGVを起動してFile => Open Sessionを選択、

カスタムした設定で読み込まれました。ポジションも完全に再現されています。

CUI環境でIGVを起動するなら、オプションなしでxmlを指定します。
igv igv_session.xml
(前回と同じパスから起動する)
3、配列をコピーする。
様々なコピー方法が用意されています。まずはリファレンスの特定の領域の塩基配列をコピーするやり方です。メニューのアイコンからDefine regionsを選択します(上辺が赤いフロッピーディスクのようなマーク)。

アイコンの形が変わるので、カバレッジウィンドウ内でコピーしたい領域の左端か右端をクリックします。

縦に線がつきました。
左端を決めたので、次に右端をクリックします。クリックすると、その領域の上に赤いバーがつきます。

赤いバーを右クリックしてコピーします。

Copy sequences を選択、
テキストエディタにペースト。コピーされています。

赤いバーを右クリックしてremove~を選択するとバーは消えます。
次はシークエンシングリードの塩基配列をコピーしてしてみます。ここでは、ソフトマスクされた領域を調べることを目的とします。まずはソフトマスク領域を表示するようにIGVの設定を変更します。
View => Preference 、出てきたウィンドウのAlignmentタブを選択します。

show soft-clipped basesにチェックを付けます。これはデフォルトではオフになっています。

saveして閉じます。
末端がソフトクリップされたリードの全長が表示されました。リードの上で右クリックしてCopy read sequenceを選べば、そのリードの塩基配列がコピーされます。

ソフトクリップされた塩基配列だけコピーしたければ、copy left-clipped sequenceを選択します(ソフトクリップ領域がないリードでは表示されない)。ここではリファレンスと異なるGTTGGTTTTという配列だけがコピーされました。

次はCopy consensus sequenceです。これを選択すると、現在ウィンドウに表示されている領域のリファレンス配列とポジションごとのアラインメントの全塩基がATGCNそれぞれ集計されてコピーされます。

テキストエディタにペースト。samtools pileupの前半のフィールドと同じようなフォーマットになります。このままRなどで分析できます。

最後です。リファレンスにない塩基が挿入されている時、その挿入配列だけをコピーします。挿入はリードの上でⅠ(アイ)として表されます。

Ⅰ(アイ)のマークの上で右クリックしてCopy insert sequenceを選択するとコピーされます。

4、表示のカスタマイズ。
標準のリードアラインメントでは画面に収まりきらいない場合、右クリックしてExpandから変更します。collapsedに変えるとリードの厚みを小さくして、より多くのリードを表示できます。Suqalishedだと全てのリードがウィンドウ内に収まるように圧縮されます。

上のメニューresize tracesボタン(Xマーク)をクリックすると、一括でこのSuqalished表示を行えます。
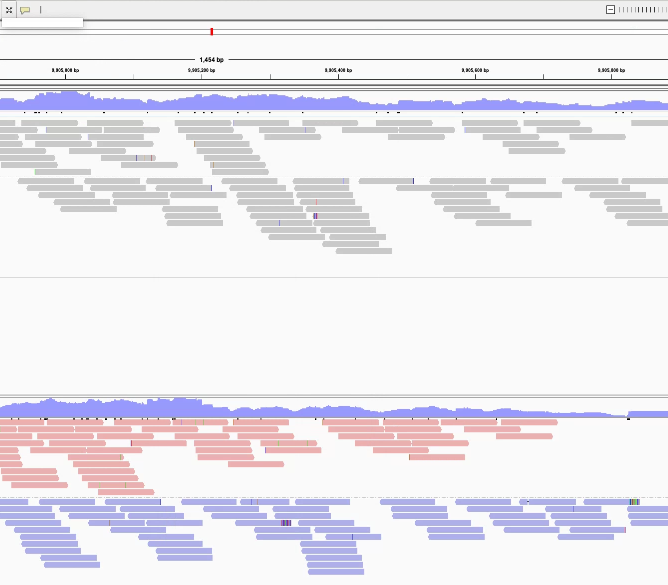
カバレッジトラックの上でshow splice junction trackを選択すると、スプライスジャンクションをブリッジのマークで表示できます。

表示されました。このshow splice junction trackも1つのトラックです。

左端のトラックをシフトキーを押しながらクリックして、全トラックを選択します。それから右クリックすると、トラックの一括設定が行えます。

change track heightで200にします。

カバレッジトラックの高さが200になりました。

リードトラックを一時的に非表示にするなら、右クリックしてShow alignment Trackのチェックを外します。

リードトラックを完全に消すなら、右クリックしてremove trackを選択します。リードトラックは完全に消えます。

いかにもjavaのGUIライクなトラック上下の二重境界線が邪魔なので、トラックをドラッグして1つのトラック内にカバレッジトラックを集めます。
移動しました。

追記
二重線を最初から非表示にしたければ、Preference => General => Display all tracks in a single panelにチエックをつけるのも手です。

色を変えます。各トラックの上で右クリックしてchange track colorを選択します。
変更しました。

右クリックしてsave imageを選択するとウィンドウ画像を保存できます。

View => set name panelを選択して、ネームパネルの幅を広くします。

ここでは300を選択。

続いてchange fot sizeでフォントサイズを変えます。

ここでは30を選択、

大きくなりました。

色々カスタマイズしたら、セッションを保存しておきます(2参照)。
引用
Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration
Helga Thorvaldsdóttir, James T. Robinson, Jill P. Mesirov
Brief Bioinform. 2013 Mar; 14(2): 178–192. Published online 2012 Apr 19
参考
関連