ゲノム編集後の突然変異の結果を確立することは、高効率のゲノムターゲティングツールの出現により、ますます重要となっている。次世代シーケンサー(NGS)は、特定の標的部位における変異誘発の程度を調査するために不可欠な方法となっている。そのため、研究者がNGSデータから変異プロファイルを取得することを可能にする堅牢で使いやすいソフトウェアが必要とされている。本発表では、あらゆる標的実験(CRISPR、I-SceI、TALENsなど)のシークエンシングデータを、欠失、一塩基変異、(テンプレート)挿入、タンデム重複などのイベント分類に焦点を当てて解析できるツール、Sequence Interrogation and Quantification (SIQ) を紹介する。SIQの結果は、SIQPlotteRという対話型ウェブツールで直接分析・可視化することができる。SIQは、様々なDNA修復不全の遺伝的背景から得られた変異シグネチャーの違いを容易に識別できることを、斬新で洞察に満ちたトルネードプロットの可視化を出力として用いて説明する。SIQは、特定の遺伝子座における変異プロファイルを確立することにより、複雑なシークエンシングデータの解釈を非常に容易にする。また、著者らの知る限り、サンガーシークエンシングデータだけでなく、イルミナやPacBioなどのショート・ロングリードNGSデータも解析できる初めてのツールである。
SIQは、例えば、生体内でヌクレアーゼによって標的とされ、その後、細胞修復経路によって修復された標的部位をカバーするPCR産物のシークエンシングによって得られるデータを利用する(論文図1A)。SIQは、それぞれが単一の変異を含むキャピラリー(サンガー)配列のコレクションを処理することができるが、SIQの真の強みは、変異の結果を広範囲に混合し、NGSにより深くシークエンシングされたプールDNAの変異プロファイルを特定できることである(論文図S1)。ショートリードシーケンス(イルミナペアエンドシーケンスなど)が適用される実験のSIQ解析では、リードがある程度重複するように、<290bp(2×150bpペアエンドリード)または<580bp(2×300bpペアエンドリード)のPCRアンプリコンを使用することを推奨する。ロングリードの場合、これらの基準は適用されず、より大きなアンプリコンが使用できる(例:>3kb)。
利用可能なデータ(Githubより)
- Sanger sequences (.ab1 files) (Note: only Sanger sequences containing a single mutation can be analyzed)
- Illumina single and paired-end sequence data (.fastq or .fastq.gz files)
- PacBio data (.fastq or .fastq.gz files)
インストール
ubuntu20.04にて、リリースからv1.1をダウンロードしてテストした。
依存
- Java version (1.8 and up).
wget https://github.com/RobinVanSchendel/SIQ/releases/download/v1.1/SIQ_1.1.jar
立ち上げる。
> java -jar SIQ_1.1.jar
(論文より)
初期チェックとして、SIQはすべてのファイルが配置可能かどうかを確認する。さらに、ユーザーは、予想される標的部位(例えばCRISPRカット部位)を定義するフランク配列を定義できる(強く推奨される)。左側と右側のフランク配列の中間が予想されるターゲット部位となり、その位置は0に設定される。提供されるフランクは≥15bpでなければならず、参照配列に存在することが要求される。2つのターゲットが使用される場合(例:2つのsgRNAが使用される場合)、フランク配列を分離することができる:左フランク配列の終わりは1つのターゲットサイトを定義し、右フランク配列の始まりは第2のターゲットサイトを定義する。実験に使用するプライマーも提供することができ(推奨)、参照DNA配列にも存在する必要がある。プライマー配列は、リードが定義されたプライマー内で開始することを確認するために使用される。R1 および R2 NGS ファイルが提供された場合、SIQ は Flash (v2.2.00) を使用してペアエンドデータのマージを試みる。その後、SIQはマージされたファイル(R1が提供された場合はR1のファイルのみ)を使用してマッピングを行う。ショートリードの場合、最初にリードの向きを確認し、同じ向きが使用されているか判断する。PacBioのデータでは、リードの向きは、リードによって、順方向と逆相補方向の両方が使用される。塩基品質の閾値以下の塩基はカットされ、高品質のリードが残される。このリードは、提供されたリファレンスにマッピングされる。ショートリードシーケンスでは、リードはプライマー結合部位内から始まり、検出されるイベントはプライマー結合部位から少なくとも5塩基(オプションで設定可能)離れたところから始まる必要がある。これにより、プライマーがDNAの意図した位置でアニーリングした場合にのみ変異原性イベントが検出されるようになる。最後に、SIQは、提供された参照配列との差に基づいてリードを分類し、Excelの表を出力する。
Example dataをロードした。リファレンスとシークエンシングデータ(ペアエンドfastqとシングルエンドfastq)のパスが表示されている。
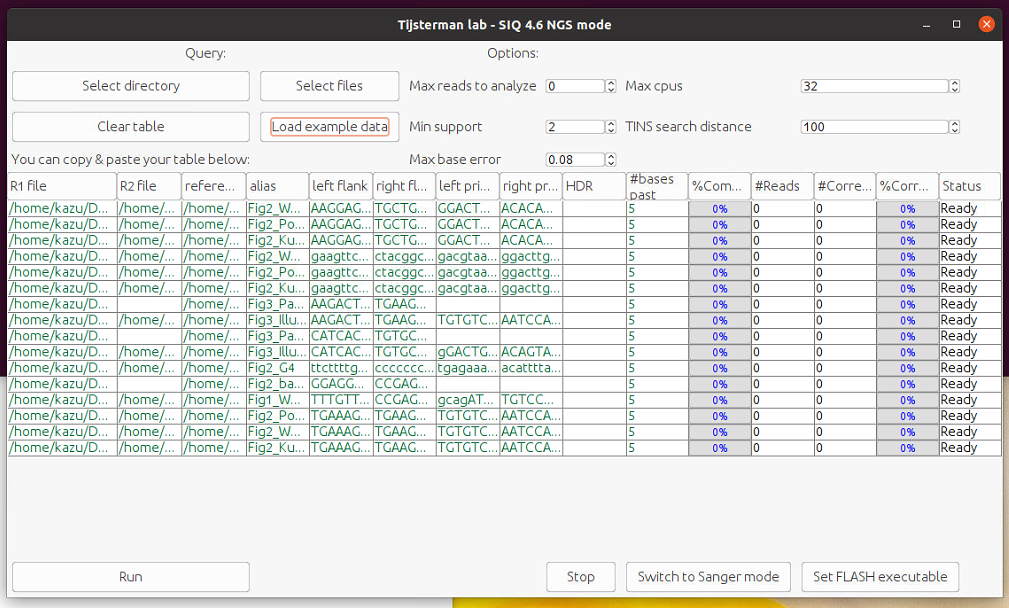
Runボタンをクリック。右上のパラメータ設定で計算がスタートする。それぞれゲージが100%になったら終了。
全ての計算が終わると出力ディレクトリが表示される。

SIQの出力はExcelの表で、直接分析することも、SIQPlotteRという専用のウェブツールで視覚化して分析することもできる。
SIQ.xlsx

SIQPlotteR (shiny web)
https://siq.researchlumc.nl/SIQPlotteR/にアクセスする。

SIQで得られたエクセルファイルを指定する。
ロードされた。
トルネードプロットでは全ての変異の種類と重みをまとめて表示する。

それぞれの色はイベントの種類を表し、各色の高さは全体の割合への寄与を表している。横軸はポジションを表す。空白は各イベントの欠失サイズを表す。欠失の場合は、接合部に見られるマイクロホモロジーの程度や挿入の有無に基づいて、さらに色分けが加えられている。このように、その増幅領域における全ての変異を視覚化できるようになっている。
表示する変異の種類はメニューのmutation typeから選べる。

このパネルは全てのイベントを示している(黄緑がWT)。一番下のwild-typeを外せば、WTイベントを除くパネルも表示できる。
複数の遺伝子の結果が読み込まれた場合は、遺伝子を選択できる。

複数のサンプルの結果が読み込まれた場合は、サンプルを選択できる。下の画像では4サンプル全てが表示されている。

Mutation types
各サンプルについて、突然変異のタイプが相対的割合か絶対的割合のどちらかで表示される。下の画像では4サンプルの結果が表示されている。

Homology plot

Size Plot

SNVs

Target Alteration plot

Sample Information plots

Tornado Plot - Templated Insertions

引用
SIQ: easy quantitative measurement of mutation profiles in sequencing data
Robin van Schendel, Joost Schimmel, Marcel Tijsterman
bioRxiv, Posted April 10, 2022
関連