JBrowse 2 desktopはシークエンシングデータのマッピングファイルの読み込みに対応しており、リファレンスゲノムに沿ったリードのアラインメントを表示することができる。
読み込み可能なデータ。ファイルの種類によってはインデックスも必要。
- Tabixed VCF
- Tabixed BED
- Tabixed GFF
- BAM
- CRAM
- BigWig
- BigBed
- .hic file (Juicebox)
- PAF
(マニュアルより)
bamファイルを読み込んでみる。
File => Open Trackを選択

右端にAdd a trackパネルが表示される。

4つボタンがあり、左端のボタンはローカルのファイルの指定、左から2つ目のボタンはファイルのURLの指定となる。右2つのボタンはdropboxとGoogle driveのファイル読み込みのボタンとなる(共有設定をONにしてURLを指定する)。ファイルの種類によっては、インデックスも提供する必要がある(画像2段目)。

NEXTをクリック。
続いて、トラックの名前、ファイルの属性を指定する。名前はsample1_chr21_mappingとした。
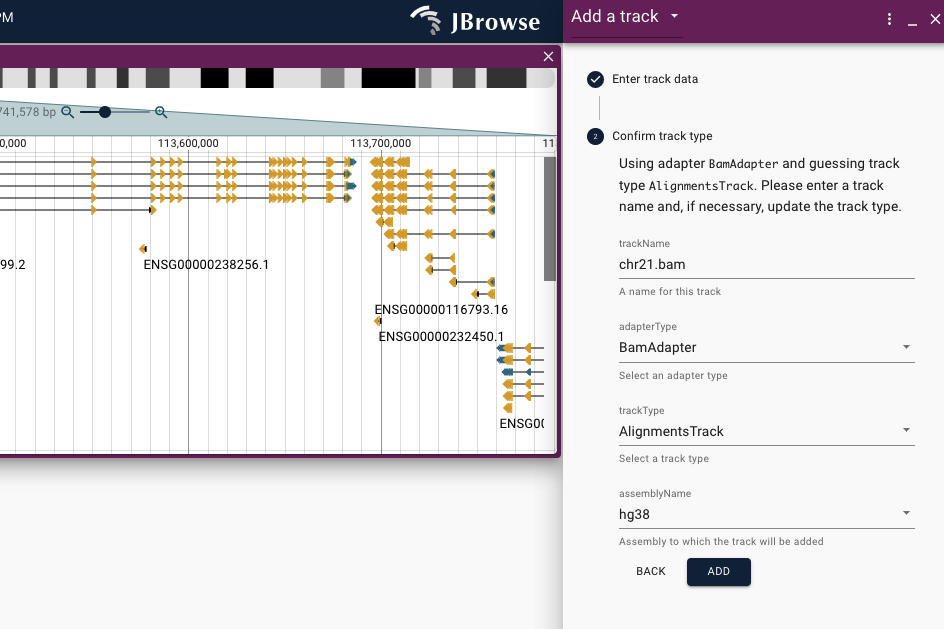
adapterTypeはファイルタイプによって変える。bamファイルならBamAdapterを指定。

trackTypeもデータのタイプによって変える。bamファイルならalignment trackを指定。

ADDボタンをクリックするとTrackがロードされる。
染色体を変えるには中央上のポジションのウィンドウをクリックする。

ロードされた(データはNA12878)。リファレンスゲノムの順鎖にアラインしたリードは赤色、逆鎖にアラインしたリードは青色で表示される。ミスマッチは色付きで描画される。

alignment trackはカバレッジとパイルアップ(リードの積み重ね)に分かれている。
ロングリードのマッピングファイルも表示できる(データはNA12878)。

リードをクリックすると、そのリードの配列やマッピングの詳細(tagやFLAGなど)が表示される。

リードの上で右クリックすると、リードの詳細を全てコピーできる。

リードの上やカバレッジトラックの上にマウスカーソルを合わせて停止すると、詳細が表示される。

Insertion
紫色は挿入、青色はソフトクリッピング、赤色はハードクリッピングをそれぞれ表す。

リードの30%をカバーしていると色のついた逆三角形でマークされる。10bp以上の挿入は、特別に大きな紫色の長方形で表示される。
Deletion

左上のマークをクリックしてshow center lineにチェックを付ける。

中央線が表示された。

トラックタイトル右端のマーク”⁝”(トリコロン、三点リーダ)をクリックすると、トラックをカスタマイズできる。

show soft clippingをオンにした。

ソフトクリップされたリードの領域も表示された。

カラースキームをPer-base qualityに変更。

カラースキームをStrandに変更。IGVと同じ配色になっている。

カラースキームをStrandに変更後、sortでRead strandを選択。

ソートのbase pairは特定のバリアントを持つリードがソートされるように積み上げられたリードを並べ替える。この機能は中心線がある部分のバリアントがソート対象になる。

また、アラインメント情報を示すタグでソートやフィルタリングしたり、色分けできるようになっている。
sort by tag

マニュアルでは、タグを利用して塩基の修飾に色を付ける例が説明されています。
現在画面に表示されている結果はSVG形式でダウンロードできる。アプリ左上のボタンをクリックしてExport SVGを選ぶ。

左上ボタンをクリックしてHorizontally flipを選ぶと染色体が反転する。反転した状態だと、座標を左から右ではなく右から左に移動させることができる。もう一度選ぶと元に戻る。

どう使うかだが、逆位などの複雑な構造多型を解釈しやすくするために便利かもしれない。
ロングリードを右クリックしてDotplot of read vs refを選択すると、リードとリファレンスのdot plotを表示できる。

linear dot plotビューが下のパネルで開いている。
同じく、ロングリードを右クリックしてLinear read vs refを選択すると、リードとリファレンスのシンテニーマップを表示できる。

これらの機能は構造多型が発生している領域の分析に有用だと思われる。
構造バリアントコール検査用に、SV inspector という機能も用意されている。
ADD => SV inspectorを選択

SVやCNVのファイルを読み込む。VCFまたはVCF.gz、BEDPE、STAR-fusionの結果ファイルを指定できる。

ここではcuteSVによるSVコール結果を選択した。
ロードされた。左にファイルの各行を表すテーブルビュー、右側にSVの環状全ゲノム表現での概要が表示される(circosビュー)。

テーブルは、検索とフィルタリングが可能。フィルタリングと検索の結果は環状ビューにも反映される。
環状ビューでフィーチャーをクリックするか、テーブルの左端の列にある三角形のドロップダウンをクリックすると、"スプリットビュー" または "ブレークポイントスプリットビュー" と呼ばれるデータの新しいビューが表示される。この機能によって、染色体間転座などの構造バリアントのブレークポイントを表示し、その部位の遺伝子アノテーションを読み込めば、SV発生部位にどのような遺伝子が存在しているか素早く確認することが可能になる。

マニュアルより転載
1000genome VCFのようなマルチサンプルVCFのポジションは1つで表示される(IGVとは異なる部分)。コール部位をクリックすると右下側にサンプルトラックが表示され、どのサンプルのコールか確認できる。

サンプルトラックではフィルターをかけて絞り込むことができる。プレーンテキスト検索または正規表現形式の検索を使用できる。 genotype フィールドに 1 と入力すると、0|1 または 1|1 のような最初の代替アレルを含むすべての genotypes が検索される(ツールの説明)。

マニュアルでは、bigwigの可視化、Hi-C データの可視化、全ゲノムスケールのCNVビューの表示方法などについても説明されています。アクセスしてみて下さい。
引用
JBrowse: a dynamic web platform for genome visualization and analysis
Robert Buels, Eric Yao, Colin M. Diesh, Richard D. Hayes, Monica Munoz-Torres, Gregg Helt, David M. Goodstein, Christine G. Elsik, Suzanna E. Lewis, Lincoln Stein & Ian H. Holmes
Genome Biology volume 17, Article number: 66 (2016)
関連