2022/11/26 追記
SRA Run Selectorは、SRAに保存されている大規模なランのセットを取り出し、どのランを解析に使用するかを絞り込み、結果をメタデータとしてダウンロードすることができる。
NCBI-Hackathons/SRA Run Selector Tutorial
NCBI Minute: Using the SRA RunSelector to Find NGS Datasets
試してみる。biofilmのメタゲノムデータセットを探す。NCBI SRAでbiofimとタイプ。
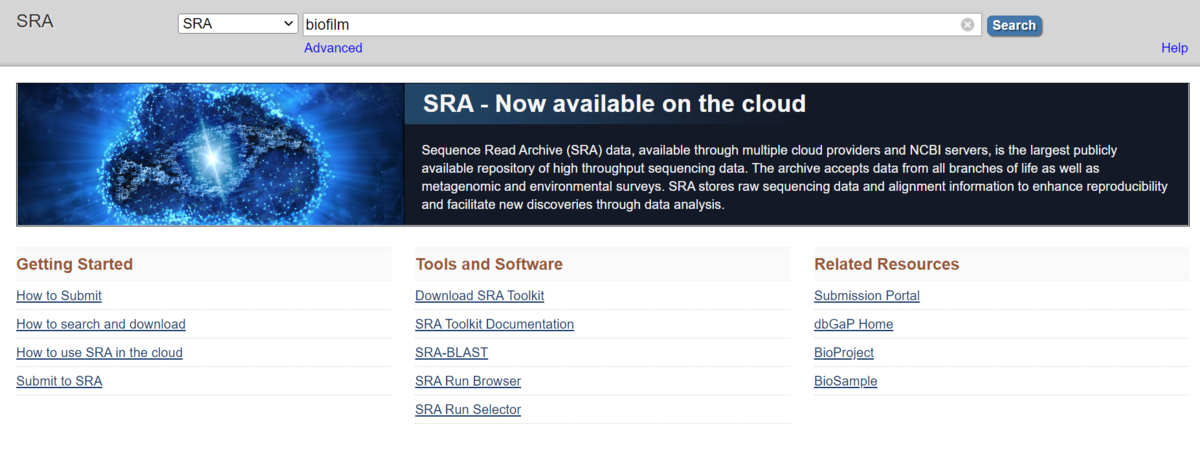
56032ヒットした。
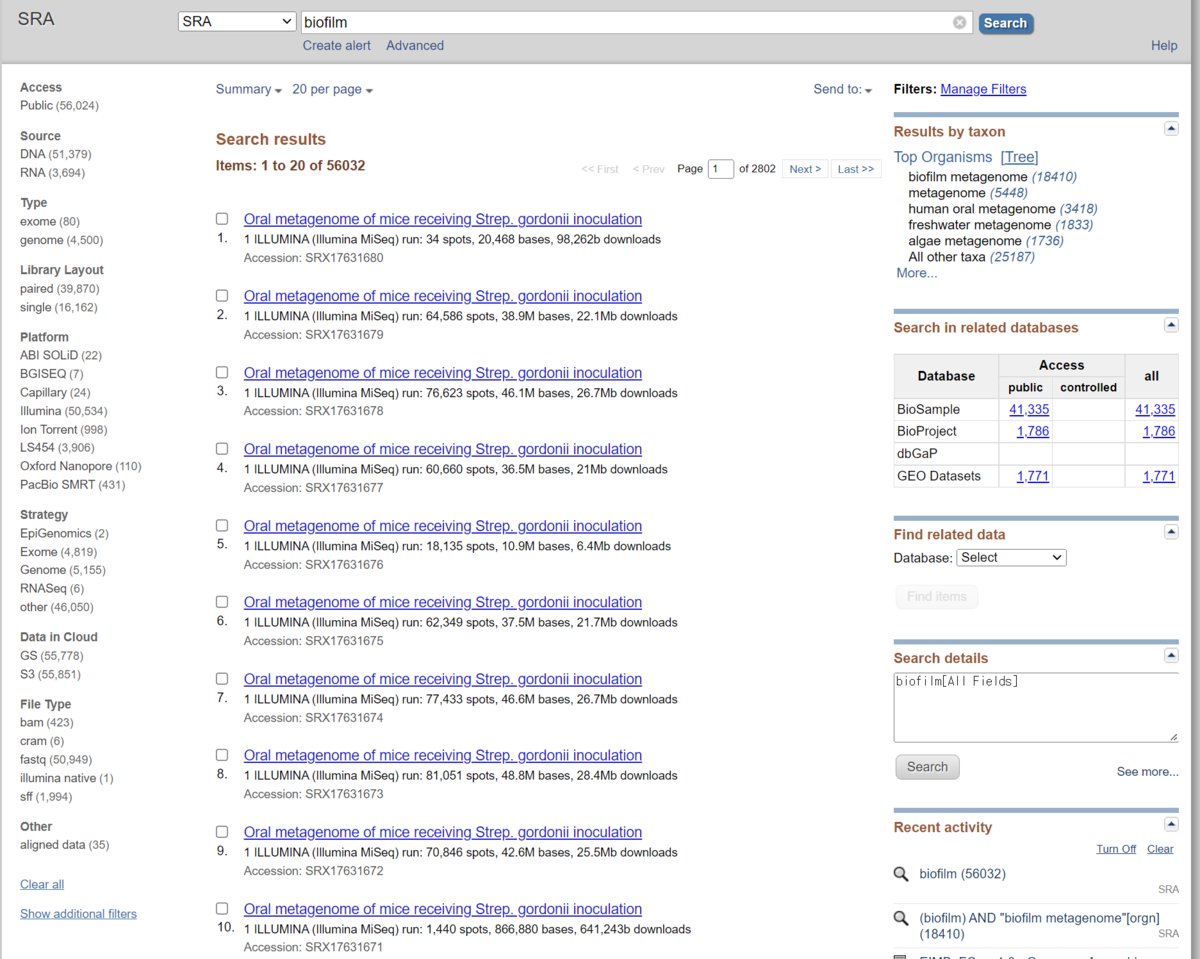
右上のtop organismでは、metagenomeの検索ではあるが、biofilm metagenomeというtaxonomyが18410ヒットしている。ほかにも口腔biofilm(プラーク?)などが出てきてるが、自然環境のデータに興味があったので、biofilm metagenomeをクリックした。"metagenome"を選んだので、biofilmだけではたくさんヒットしていたかもしれないメタアンプリコンのデータは効果的に除外されるはずである。
結果。まだ絞れそうだが、Send results to Run selectorが出てきたのでクリック。
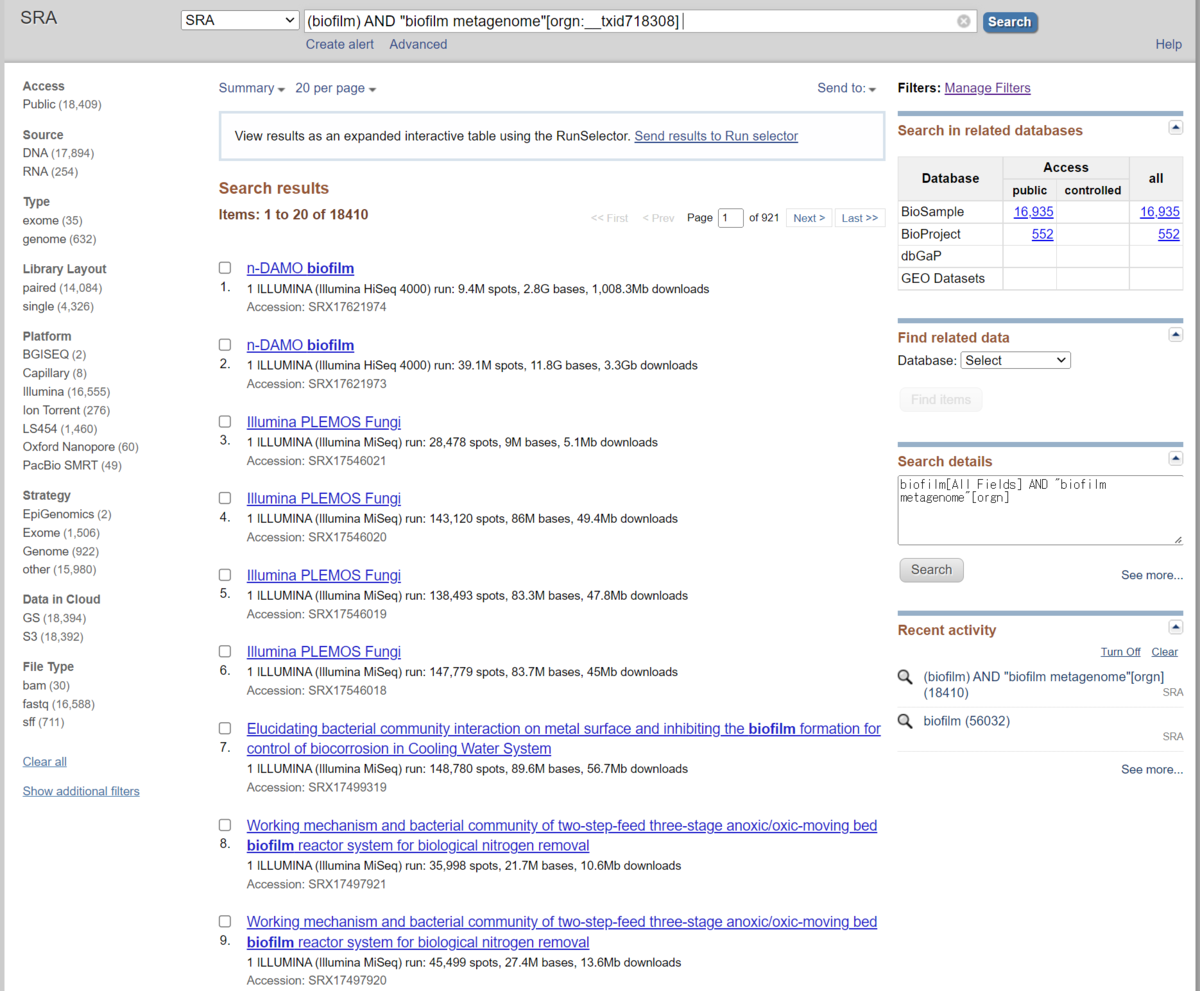
このSend results to Run selectorは、検索結果の画面で一度でも絞り込みを行うと出てくる。
追記
SRA Run selecterは数が多すぎると使用できないようなので、フィルタだけで絞り込めない時はNOT検索も併用する。効果的に絞りこみできる。soil metagenomeのNOT "amplicon"で40万=>3万まで減った。
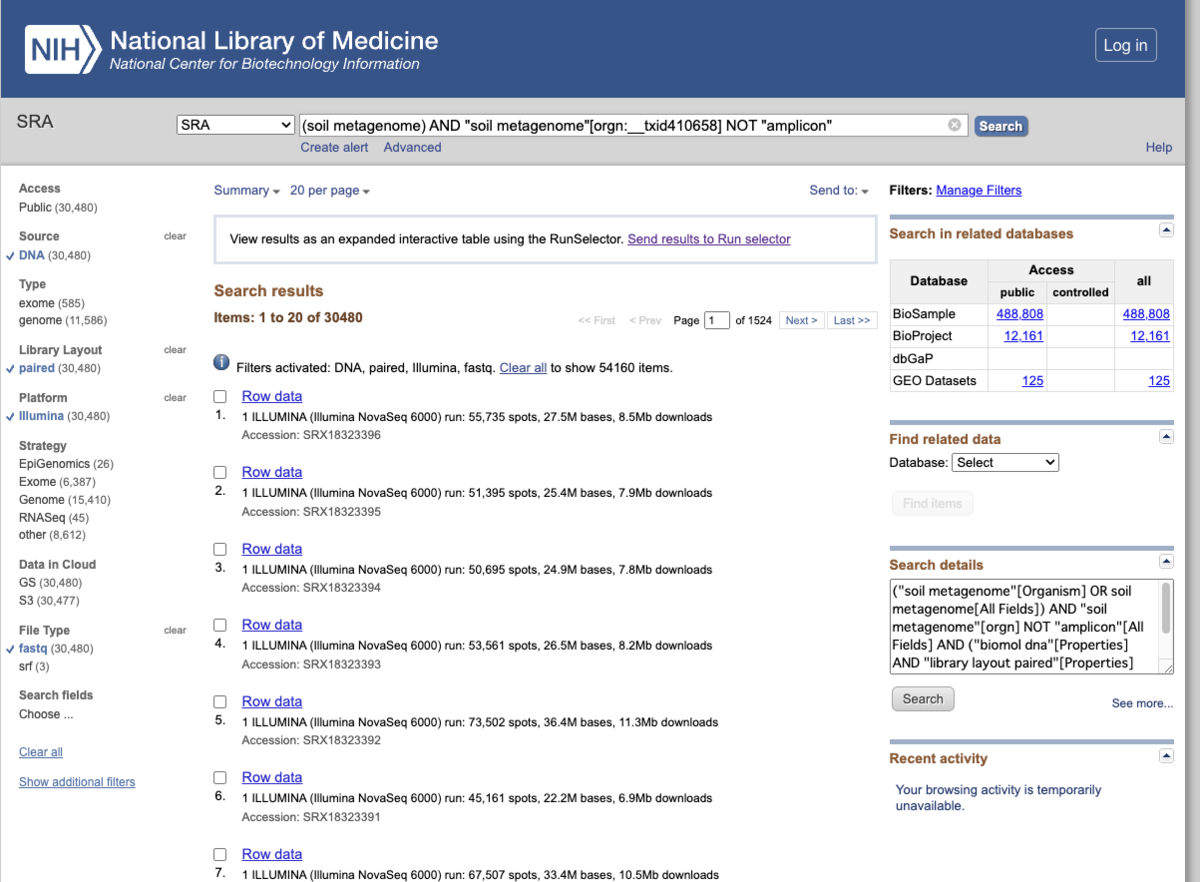
Run selectorに移動した。
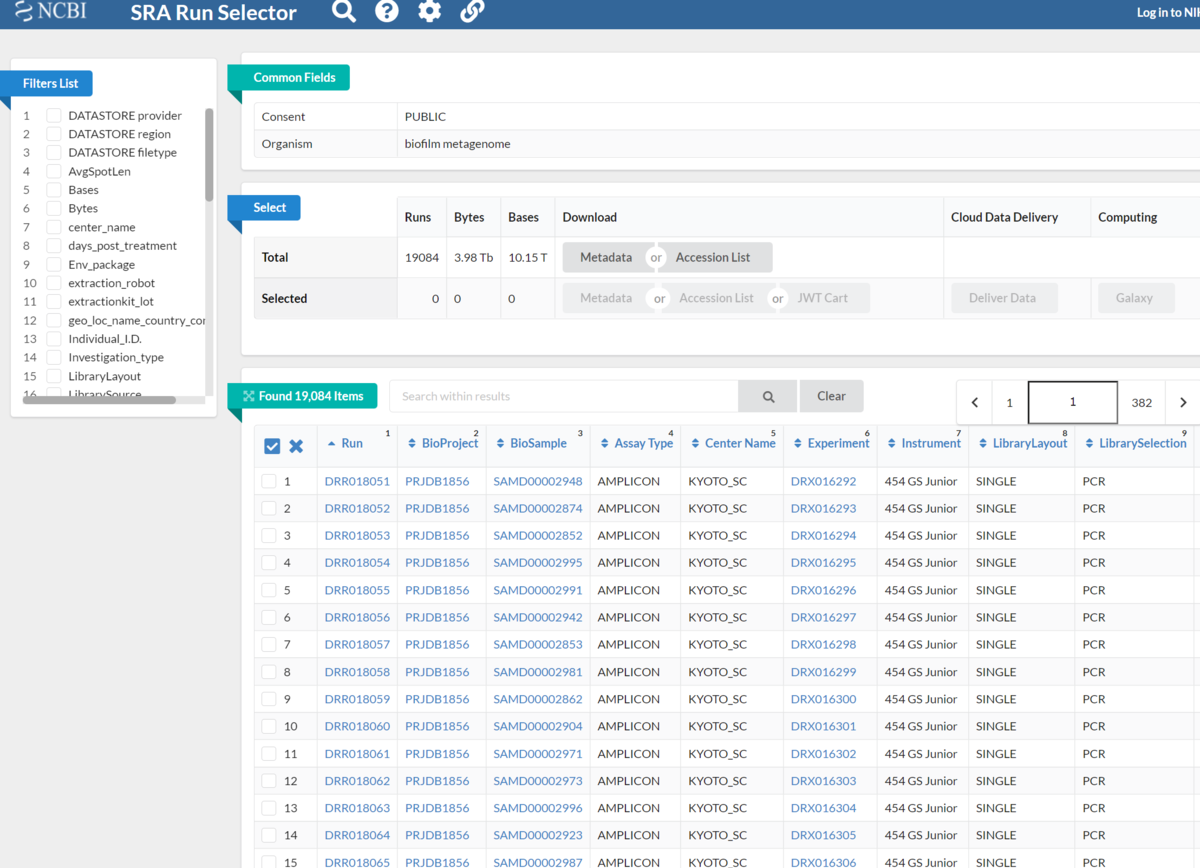
注;dbGaPのデータは許可されているアカウントでログインしていないと扱えない。また、アクセスする前にコールドストレージから取得する必要があるものもある。その場合、クラウドベースのコールドストレージを経由して転送することになる。
Run selectorでは、関心のあるRunデータを絞り込むために様々なフィルターを適用できる(表示されるフィルターはリストによって変化する)。ここではPlatformをクリックした。

Platfoprmが表示された。ILLUMINAが多いが、他のシークエンスプラットフォームも見つかる。ILLUMINAを選択した。
ILLUMINAの16666ランに絞り込まれた。
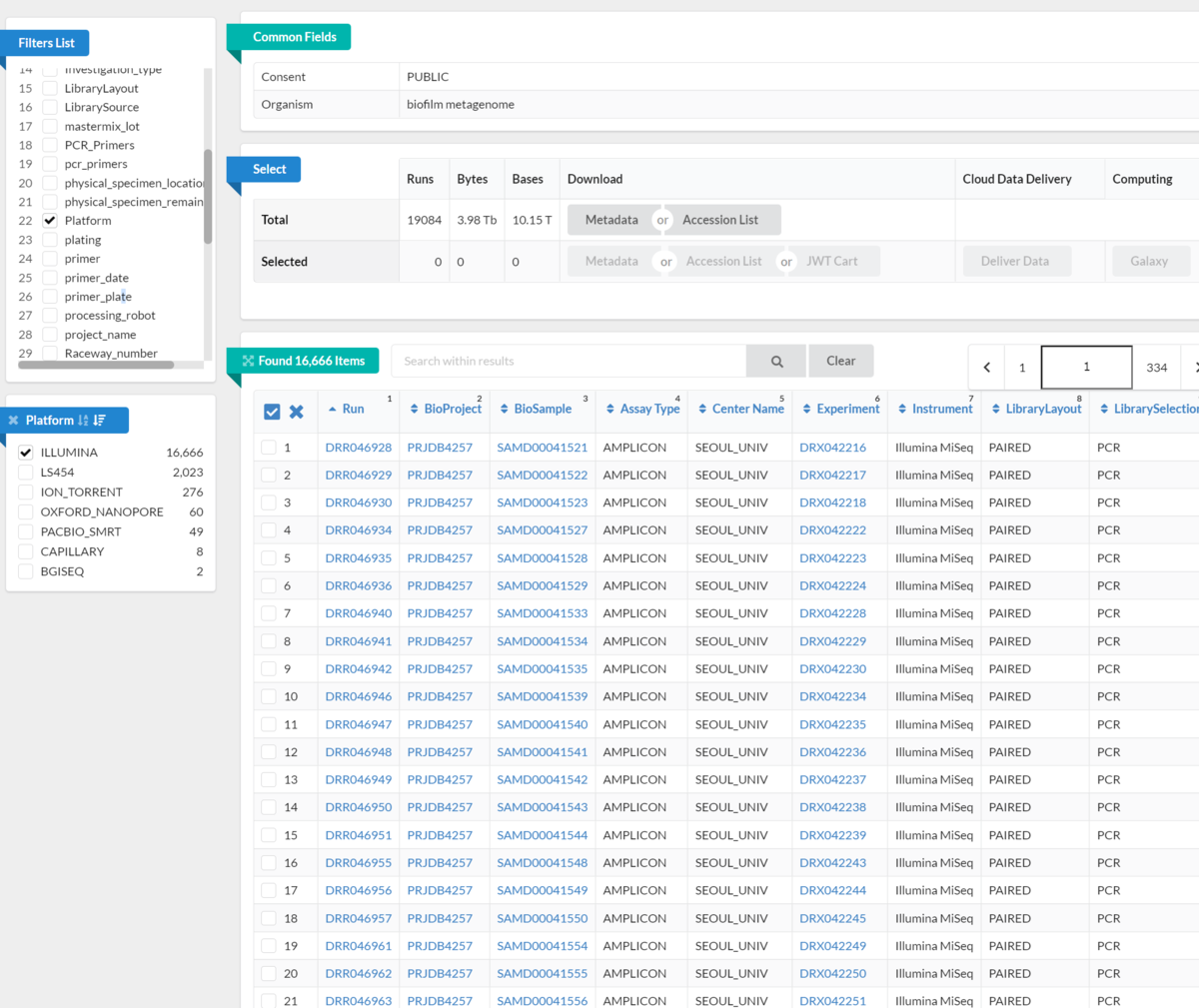
(フィルターは複数重ねて適用できる。)
ランのチェックをつけると、直接絞り込むことができる。下の写真のように4つだけ選択してみた。選択したデータの個数と総塩基数などが写真上部のSelectedに表示されている。Bytesが194.24Mb、Basesが219.70 Mとなった。

今回は絞り込んだ16666ランのデータをダウンロードするため、Xマーク左をクリックして全部選択する。(Xマーククリックで解除)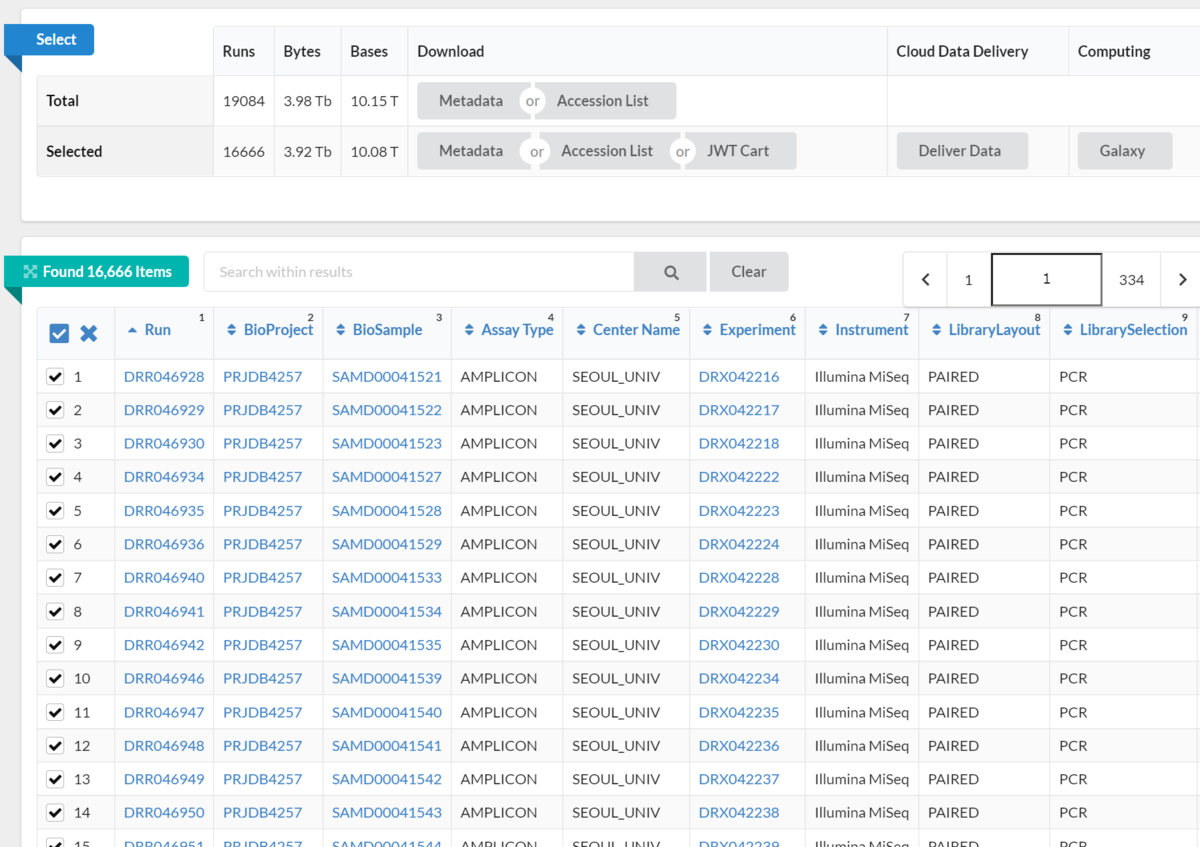
全部選択するとSelectedが16666となった。
SelectedのMetadataをクリックするとメタデータテーブル全体がダウンロードされる(間違えて絞り込む前のAllの方をクリックしないように注意)。

SlectedのAccesion listをクリックするとIDだけがダウンロードされる。

リストが手に入ったら、fastq-dumpを使ってデータを取得する。ここでは目的の細菌種が決まっているので、そのようなデータが含まれるのか調べるために少数だけリードを取得する。数が多いと時間がかかるので、GNU parallelで10並列し、30万リードずつだけ取得する。-Nと-Xはイルミナフローセルの座標に相当する(スポットの指定)。0から取得するのは品質が問題になる気があする?ので 10,000から開始して310,000まで。
#--dry-runを付けてチェック。OKなら外す
cat Accesion_list| parallel -j 10 --dry-run 'fastq-dump --split-files -N 10000 -X 310000 --gzip {}'
- -N Minimum spot id
- -X Maximum spot id
- --split-files Write reads into separate files. Read number will be suffixed to the file name. NOTE! The `--split-3` option is recommended. In cases where not all spots have the same number of reads, this option will produce files that WILL
CAUSE ERRORS in most programs which process split pair fastq files. - --gzip Compress output using gzip: deprecated, not recommended
1000サンプルで2~3時間ほどかかった。
結果はkraken2などで分析する。
#!/bin/sh
for file in `\find *_1.fastq.gz -maxdepth 1 -type f`;
do
fastq1=$file;
fastq2=${file%_1.fastq.gz}_2.fastq.gz;
kraken2 --db <path>/<to>/kraken2-database --threads 20 --gzip-compressed --paired --report ${file%_1.fastq.gz}_kraken2_report.txt --use-names --classified-out seqs#.fq $fastq1 $fastq2 > ${file%_1.fastq.gz}_output.txt;
rm seqs_1.fq seqs_2.fq ${file%_1.fastq.gz}_output.txt;
bracken -d <path>/<to>/kraken2-database -i ${file%_1.fastq.gz}_kraken2_report.txt -r 100 -l S -t 5 -o ${file%_1.fastq.gz}_bracken;
python KrakenTools/kreport2krona.py -r ${file%_1.fastq.gz}_kraken2_report_bracken.txt -o ${file%_1.fastq.gz}_bracken_convert;
done
#kronaで可視化
ktImportText *_bracken_convert -o all.html
NCBIの動画でわかりやすく説明されています。視聴してみてください。ただ2017年の動画でインターフェイスが古いものになっています。
引用
参考
Biostars
https://www.biostars.org/p/12047/