2021 1/8 誤字修正
大規模な並列シーケンシングの進歩とシーケンシングコストの劇的な削減により、RNAのディープシーケンシング(RNA-Seq)はRNA転写産物の同定と定量化のための主要なツールとなった。今日、RNA-Seqは、創薬標的の同定、新規遺伝子制御機構の予測などを目的とした研究において、細菌の遺伝子発現を解析するために広く利用されている。このような研究では、多くの場合、計算データの取り扱いと生物学の両方の深い知識が必要とされる。バイオインフォマティクスの中程度の知識しか必要としないスタンドアロンのパイプラインやツールがいくつかあるが、原核生物のRNA-Seq解析は、利用可能なほとんどのRNA-Seqパッケージは、入力データが原核生物のものとは多くの面で異なる真核生物の遺伝子構造を反映していることを前提としているため、困難である(Johnson, et al., 2016)。バクテリアの転写産物は、イントロンを持たず、スプライシングされていないため、スプライスジャンクションを考慮すると誤って割り当てられたリードが増加することが多い(Magoc, et al., 2013)。さらに、真核生物とは異なり、特定のストレス下では、ほぼすべての原核生物遺伝子の発現が変化する可能性がある(Creecy and Conway, 2015)。さらに、実験セットアップのばらつき、プラスミド遺伝子の存在および過剰発現、ならびにRNA-Seqプロトコルの違いのために、原核生物のデータには、品質トリミング、アダプター除去、および歪んだデータの正規化がしばしば必要とされる(Magoc, et al.、2013; McClure, et al.、2013)。
原核生物用には、SPARTA(Johnson, et al. 2016)、EDGE-pro(Magoc, et al. 2013)、およびRockHopper(McClure, et al. 2013)のような、RNA-Seqデータの解析を容易にすることができるいくつかのソフトウェアパッケージがあるが、いずれも、データ操作に関する実質的な知識を必要とする。そこで、原核生物のRNA-Seqデータ解析を行う際の人の介入を減らすために、利用可能な様々なツールとPythonで書かれた組み込み関数を統合し、完全自動化されたコマンドラインベースのワークフローであるProkSeqを開発した。ProkSeqは、ショートリードアライナーであるbowtie2(Langmead and Salzberg, 2012)をデフォルトパラメータとして統合しているほか、擬似アラインメントのオプションとしてSalmon(Berghoff et al., 2017)も統合している。prokseqは、サンプル内およびサンプル間比較のための正規化された発現値、不要な変動(RUV)を除去するためのオプション(Risso, et al. さらに、ProkSeqは、下流のGene Ontology(GO)(Gene Ontology, 2008)およびKEGGパスウェイエンリッチメント解析(Kanehisa and Goto, 2000)をサポートしている。ProkSeqは、RNA-Seqデータの品質管理ステップから、異なる発現を持つ遺伝子のパスウェイエンリッチメント解析までを処理する(図1)。差分発現、正規化発現、可視化のオプションが豊富に用意されており、図を作成することができる。人の介入が少なく、マルチスレッド機能を備えているため、データの再フォーマットが必要となることが多い別のツールを逐次適用するよりも、ProkSeqの利用にかかる時間を短縮することができる。
ProkSeqはLinuxベースのコマンドライン環境で動作し、ユーザー定義のパラメータとサンプルファイルに依存する。パイプラインで使用されるツールはデフォルトのパラメータで設定されている。しかし、より高度なユーザーは、ツールのパラメータを調整して機能を制御することができる。サンプルファイルは、分析に含めるfastqファイルの名前を示し、また、処理群サンプルやコントロールサンプルなどの実験クラスを定義する。ProkSeqは、まずリードの品質をチェックし、FastQC(http://www.bioinformatics.babraham.ac.uk/projects/fastqc)とafterQC(Chen, et al)、シングルエンドとペアエンドの両方どちらでもbowtie2とそのデフォルトのパラメータを使用して、リードをリファレンスゲノムにマッピングする。ProkSeqはその後、各ライブラリのアライメント品質に関するレポートを図とテキストの両方で生成し、カバレッジの均一性、タンパク質コーディング配列に沿った分布、5'および3'UTR領域、ならびにRSeQCによって生成されたリード重複率とストランド特異性に関する情報を提供する(Wang, et al., 2012)。遺伝子あたりの総リード数はfeatureCounts(Liao, et al., 2014)を用いて計算される。また、各遺伝子の正規化された遺伝子発現値を、100万あたりの転写産物(TPM)と100万あたりのカウント(CPM)の形で計算する(Wagner, et al. これらの計算式については、論文の補足方法(S1)で説明している。
ProkSeqは、DESeq2(Love, et al., 2014)、edgeR(Robinson, et al., 2010)、およびNOISeq(Tarazona, et al., 2015)などの発現変動を解析するためのいくつかのツールを統合している。DEGsの下流解析にはGOエンリッチメントを使用し、clusterProfilerを統合してパスウェイエンリッチメントを行う(Yu, et al)。アライメント前後の品質統計やグラフィカルな可視化に関するレポートはpdfとHTML形式で作成される。ProkSeqの重要なユニークな機能の1つは、歪んだデータのためのRUV正規化および平均ヌクレオチドカウント法の統合である(Creecy and Conway, 2015; Zhu, et al., 2019)。さらに、このパッケージは、任意のゲノムブラウザで可視化するための1ヌクレオチド分解能のwiggleファイルを生成する。ProkSeqは、データ解析の各ステップでグラフィックスと図を生成し、ユーザーにデータへの信頼性と理解度を高める。
Documentaiton
インストール
ubuntu18.04LTSでテストした。
依存
#docker(link)
docker pull snandids/prokseq-v2.1:v1
#conda(link)
conda install -c snandids prokseq
テストラン
dockerイメージを指定して立ち上げ、続いて2行目以降のコマンドを叩いて設定を呼び出す。
docker run -it snandids/prokseq-v2.1:v1
cd prokseq
source /etc/profile.d/conda.sh
conda activate py36
以下のようなファイル構成になっている。
# ls -lh
total 111M
drwxr-xr-x 10 root root 4.0K May 29 2020 Output
-rw-rw-r-- 1 1000 1000 21K Jun 7 2020 README.txt
-rw-r--r-- 1 1000 1000 4.6M May 1 2020 SequenceChromosome.fasta
drwxr-xr-x 2 root root 4.0K May 29 2020 bowtie2_genome
-rw-r--r-- 1 root root 18 May 29 2020 chr.sizes
-rw-r--r-- 1 root root 5.0K May 29 2020 class.log
drwxr-xr-x 2 root root 4.0K May 29 2020 data
drwxr-xr-x 1 root root 4.0K May 29 2020 depend
-rw-rw-r-- 1 1000 1000 273K May 1 2020 oldAnnotationGFF.bed
-rw-rw-r-- 1 1000 1000 367K May 1 2020 oldAnnotationGFF.gtf
-rw-rw-r-- 1 1000 1000 2.9K Jun 8 2020 param.input.bowtie
-rw-rw-r-- 1 1000 1000 3.1K Jun 8 2020 param.input.salmon
-rw-r--r-- 1 1000 1000 1.3M Mar 30 2020 reads_1.fastq
-rw-r--r-- 1 1000 1000 1.3M Mar 30 2020 reads_2.fastq
-rw-r--r-- 1 1000 1000 1.3M Mar 30 2020 reads_3.fastq
-rw-r--r-- 1 1000 1000 1.3M Mar 30 2020 reads_4.fastq
-rw-rw-r-- 1 1000 1000 8.2M May 27 2020 sampleCtrl_1.R1.fq
-rw-rw-r-- 1 1000 1000 8.6M Jun 2 2020 sampleCtrl_1.R2.fq
-rw-rw-r-- 1 1000 1000 8.2M May 27 2020 sampleCtrl_2.R1.fq
-rw-rw-r-- 1 1000 1000 8.6M May 27 2020 sampleCtrl_2.R2.fq
-rw-rw-r-- 1 1000 1000 8.2M May 27 2020 sampleCtrl_3.R1.fq
-rw-rw-r-- 1 1000 1000 8.6M May 27 2020 sampleCtrl_3.R2.fq
-rw-rw-r-- 1 1000 1000 8.2M May 27 2020 sampleTreat_1.R1.fq
-rw-rw-r-- 1 1000 1000 8.6M May 27 2020 sampleTreat_1.R2.fq
-rw-rw-r-- 1 1000 1000 8.2M May 27 2020 sampleTreat_2.R1.fq
-rw-rw-r-- 1 1000 1000 8.6M May 27 2020 sampleTreat_2.R2.fq
-rw-rw-r-- 1 1000 1000 8.2M May 27 2020 sampleTreat_3.R1.fq
-rw-rw-r-- 1 1000 1000 8.6M May 27 2020 sampleTreat_3.R2.fq
-rw-rw-r-- 1 1000 1000 1.2K May 28 2020 samples.bowtie.PEsample
-rw-rw-r-- 1 1000 1000 1.1K May 27 2020 samples.bowtie.SEsample
-rw-rw-r-- 1 1000 1000 868 May 28 2020 samples.salmon.PEsample
-rw-rw-r-- 1 1000 1000 840 May 28 2020 samples.salmon.SEsample
drwxrwxr-x 1 1000 1000 4.0K Jun 8 2020 scripts
-rw-r--r-- 1 root root 18 May 29 2020 test.fasta
depend/にソフトウエアが収められている。
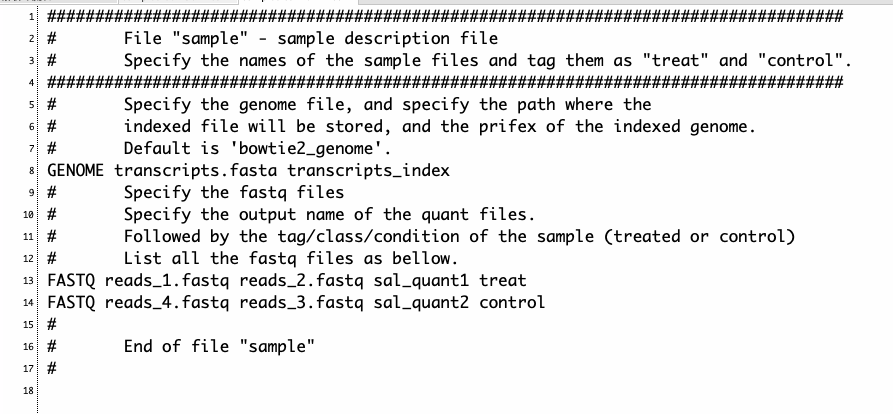
サンプル情報のシートとパラメータのconfigファイルをそれぞれ指定して実行する。テストランのサンプル情報シートは、bowtieとsalmonそれぞれシングルとペアあり、合計4種類が用意されている。
samples.salmon.SEsample

コメントアウト(#)されていない部分がユーザーが指定する部分。8行目のGENOME<space>でゲノムのfastaとbowtieのindex(bowtie2_genomeディレクトリに収納されている)を指定。13−18行目にかけて、1データずつ、FASTQ<space>pair_R1.fq.gz<space>pair_R2.fq.gz<space>name<space>control/treatで2群間比較のデータを指定する。
samples.salmon.PEsample
パラメータもbowieとsalmonそれぞれ用意されている。
param.input.bowtie

テストはデフォルト設定でランできるが、実際のランではgtfとbedファイル名を修正する必要がある。また、GO enrichment解析はTERM2GENE.csvをもとに実行される(GO databaseはdata/TERM2NAME.csv)。GO enrichment解析を実行したいなら、事前に対応表を作って置いておく必要がある(*1)。
param.input.salmon

あとはパラメータとサンプルシートのテキストファイルを指定するだけで実行できる。-n 4はCPUの指定数。もっと多くのCPUを利用できるマシンなら、増やすことで計算を高速化できる。
python scripts/pipeline-v2.8.py -s samples.bowtie.PEsample -p param.input.bowtie -n 4
- -s provide the sample description file
- -p provide the parameters defined file
- -n provide the number of processors
File (param.input.bowtie) found.
File (samples.bowtie.PEsample) found.
Checking required tools or packages:
samtools : Found
salmon : Not found
fastqc : Found
bowtie : Found
afterqc : Found
readFasta : Found
R : Found
GnBdCov : Found
FeatCnt : Found
Checking R packages:
DESeq2 : TRUE
ggplot2 : TRUE
edgeR : TRUE
NOISeq : TRUE
limma : TRUE
clusterProfiler : TRUE
apeglm : TRUE
SALMON:
salmon IS NOT FOUND!
Done with package checks. Seems all the required packages are available.
Do you want to continue? (Y/N) :
Yで実行。
テストのfastqは数 MBしかないため、1分以内に計算は終わる。
出力

長いので分けて展開。マッピングのbam,samが確認できる。QCはFastQCやafterQCなどの前処理前後のレポート。

データサイズが多くなるとこの辺りに時間がかかるかもしれない。
plots
edgeRvolcano.pdf

pca.pdf
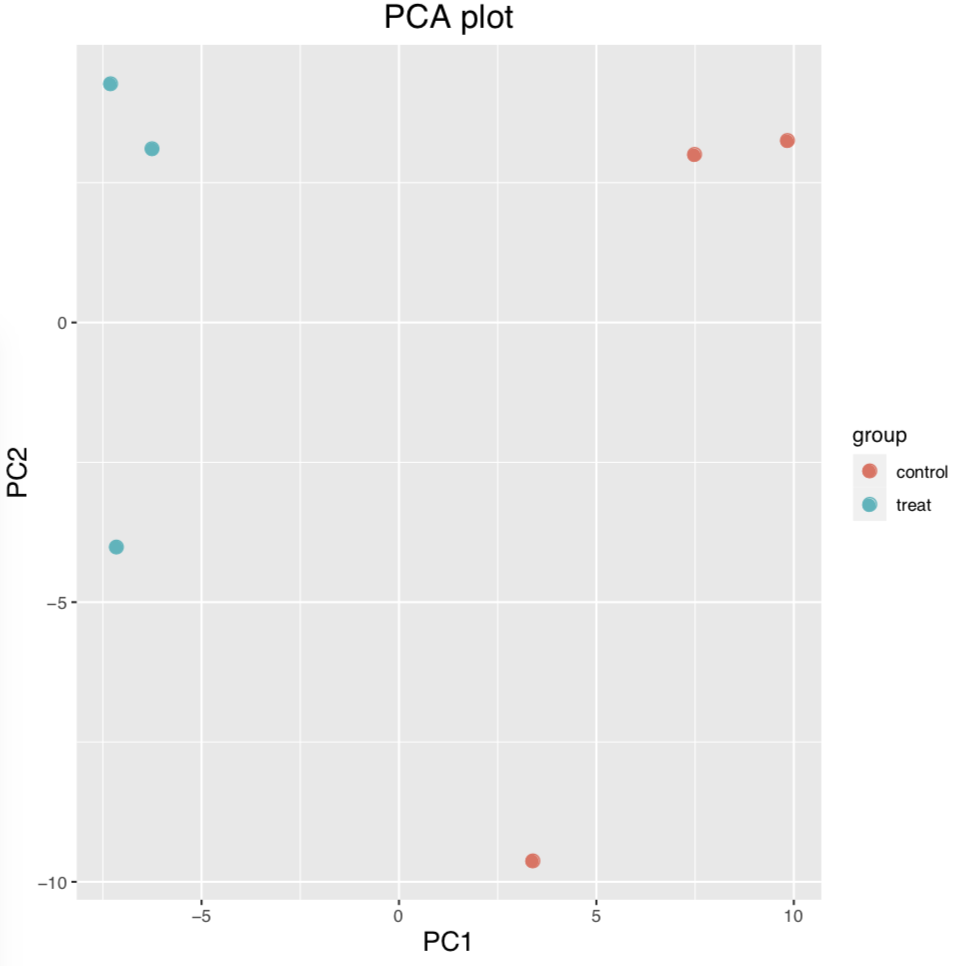
pValue-histogram.pdf
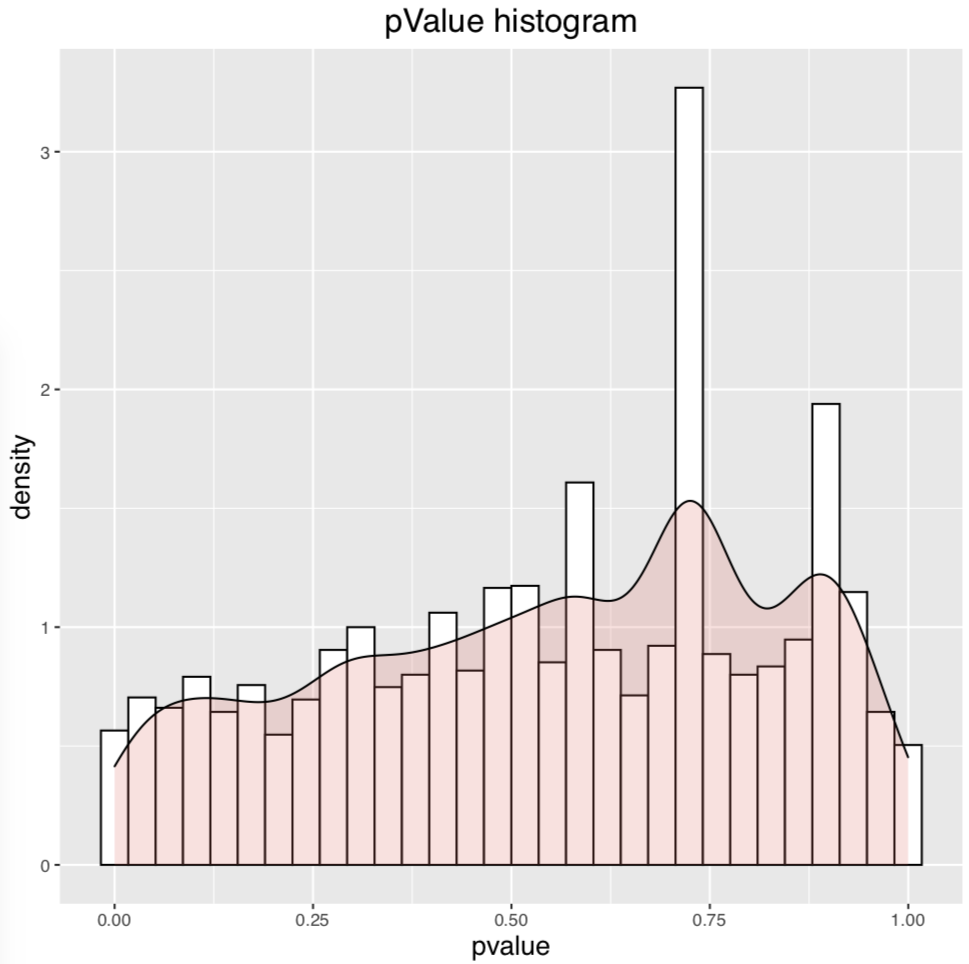 DESeq-Volcano.pdf
DESeq-Volcano.pdf

MA-plot.pdf

correlation_violin.pdf

correlation.pdf
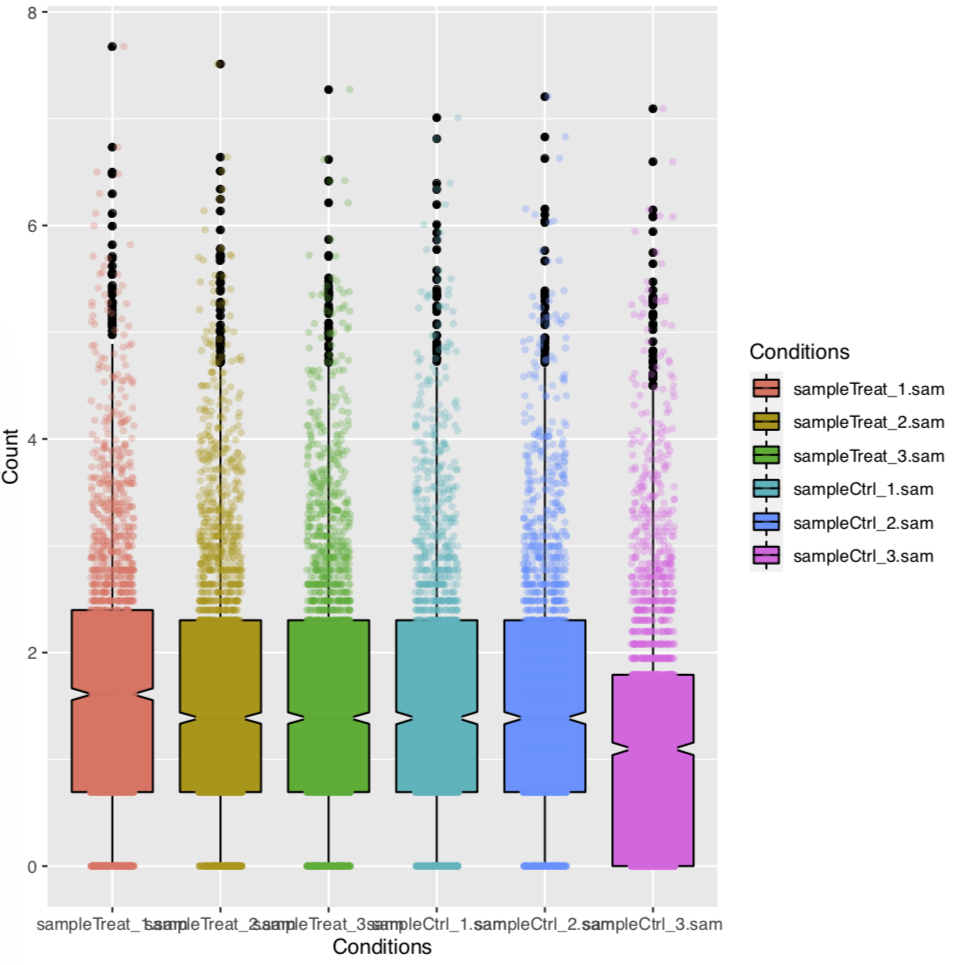
RSeQC.geneBodyCoverage.curves.pdf
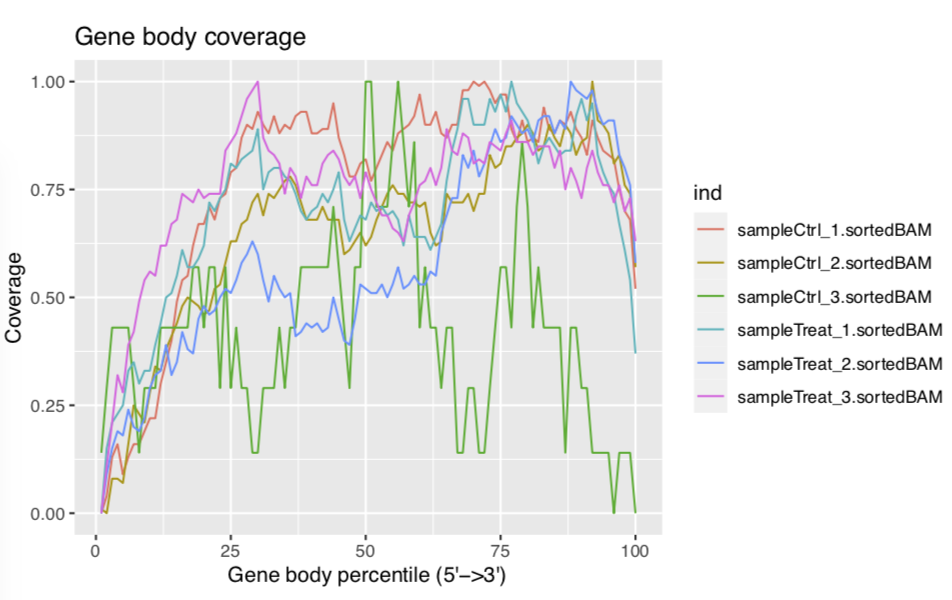
sampleCtrl_2.sortedBAM.bam_RSeQC.geneBodyCoverage.curves.pdf(この図はサンプルごとにできるのであと5つある)

AlignmentStat
countFile.NucleotideAvgCount.csv (右端にTPMがある)

Results

edgeR_results.txt

DESeq2_results.txt

DESeq2lfcShrink_results.txt

afterNoiseq.txt

GOenricher.txt

GOpathways.txt

KEGGpathway.txt

KEGGenricher.txt

実際のデータを使ったランも/root/prokseq/で行う。
cd <your>/<fastq>/
docker run -itv $PWD:/data snandids/prokseq-v2.1:v1
cd prokseq
source /etc/profile.d/conda.sh
conda activate py36
cp /data/*fastq .
#続いて編集したparameterのconfigファイル、genome、アノテーションファイルをコピー
cp /data/genome.fasta .
cp /data/index* .
cp /data/anotation* .
cp /data/bowtie.PEsample .
cp /data/input.bowtie .
準備ができたら実行する。
python scripts/pipeline-v2.8.py -s bowtie.PEsample -p input.bowtie -n 20
AfterQCなどの時間がかかるツールもあるので、実際のデータだと終わるまでに最低数時間はかかる。 まず小さな反復1で数百万リードくらいの小さめのデータセットを用意し、それで動作するか確認すること。
dockerでランする時は、データとゲノムなどを/root/prokseqに移して行う。fastqは非圧縮で提供する。
引用
ProkSeq for complete analysis of RNA-Seq data from prokaryotes
A K M Firoj Mahmud, Nicolas Delhomme, Soumyadeep Nandi, Maria Fällman
Bioinformatics, Published: 26 December 2020
プレプリントの時に一度紹介していたのですが、まとめ直しました。
参考
https://academic.oup.com/bioinformatics/article/35/12/2084/5159452
*1
TERM2GENE.csvの中身

遺伝子名は他のアノテーションファイルと合致していること。
TERM2NAME.csvはGO termを定義するファイル。
