原核生物は無性の生物であるが、これらの生物は有性生物の減数分裂による組換えとは異なり相同組換えを頻繁に行う。ゲノム進化をシミュレートするために開発されたほとんどのツールは、有性生殖を想定しているか、あるいは集団にDNAフラックスが全くないことを想定している。その結果、組換えを考慮しながら原核生物のゲノム進化をモデル化できるシミュレーターはほとんどない。さらに、多くのシミュレータはゲノム進化の中立モデルを仮定した合体論に基づいており、動物や植物のような弱い選択圧の下で進化する生物に最適である。これとは対照的に、原核生物はより強い選択圧の下で進化していると考えられており、フォワード・イン・タイムのシミュレータがこれらの生物に適していることを示唆している。ここでは、相同組換えをモデル化した原核生物のコアゲノム進化のフォワード・イン・タイム・シミュレーターであるCoreSimulを紹介する。シミュレーションは系統樹によって導かれ、コドン選択のモデルを含む様々な置換モデルを組み込んでいる。CoreSimulは、原核生物のゲノム進化に適用可能なシミュレータの限られたリストの中で、重要な追加となる柔軟なフォワード・イン・タイム・シミュレータである。
インストール
依存
git clone https://github.com/lbobay/CoreSimul.git
cd CoreSimul/
テストラン
control.txtがconfigファイルに相当する。これを編集する。
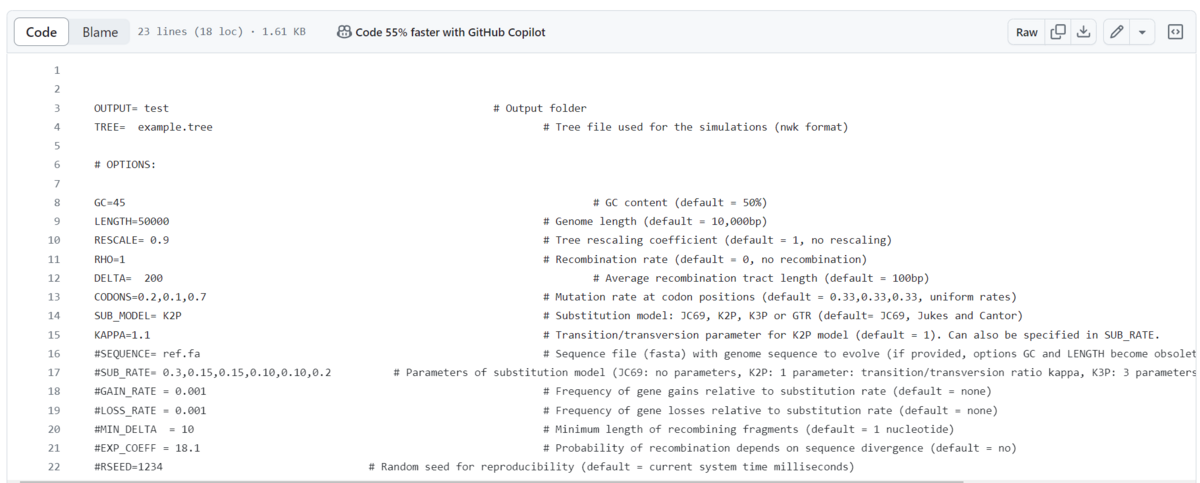
出力ディレクトリはtest。シミュレーションで使用されるツリーファイルが必要。テストのために、レポジトリにはexample.treeが含まれている。GCはGC含量、LENGTHは遺伝子長、RESCALEはツリーのスケーリング係数、RHOは組み換え率、DELTAは平均組み換え長、CODONSはコドンのポジションそれぞれの突然変異率。0.33,0.33,0.33のような一様な率ではなく、テストでは0.1,0.05,0.85と指定されている。SEQUENCEは進化させるゲノム配列ファイル(指定するとGCおよびLENGTHの設定がキャンセルされる)。KAPPAは、"K2P" モデルの遷移/転換パラメータを指定。デフォルトでは1だが、ここでは1.1に設定されている。SUB_MODELは置換モデルでK2Pが指定されている。SUB_RATEは置換モデルのパラメータで、置換の確率モデルによって指定が必要な数は異なる(塩基対の間の置換の起こりやすさを定義するパラメータ(確率)で、例えばGTRなら6つ必要)。
最後の4つはコメントアウトされているが、これらは、シミュレーションに追加の機能を提供するためのもの。GAIN_RATEは遺伝子の獲得の頻度を置換率に対して相対的に指定したパラメータ。LOSS_RATEは遺伝子の喪失の頻度を置換率に対して相対的に指定したパラメータ。MIN_DELTAは組換え断片の最小長を指定する。デフォルトでは1塩基だが、指定された場合は、組換え断片の長さがこの値よりも短い場合には組換えが発生しない。EXP_COEFFは組み換えの発生確率が配列の違いに依存する場合、その依存関係を調整する係数を指定する。これらは全てデフォルトでは無効。最後の#RSEED=1234のコメントは、指定されていない場合は現在時刻から取得されてランダムシードとするとあるが、コメントを外さないとエラーになった;変える時はnumpyで扱うseed値の32bit制限の最大の整数範囲内に収まる(2^32 - 1)ようにする。
実行する。
python coresimul_master.py control.txt
数秒かかった。
出力例
test/

CoreSimulのワークフロー(論文より)
- CoreSimulは、系統樹に基づいて原核生物のゲノムセットを生成する。CoreSimulは、コアゲノムとアクセサリゲノムの両方をシミュレートすることができる。
- CoreSimulのプロセスは、ユーザーによって指定された、長さL、GC-content のランダムなコアゲノム配列を生成すること、もしくはユーザーが提供したゲノム配列から始まる。この配列は、遺伝子間DNAを含まない、タンパク質をコードする遺伝子のヌクレオチドを連結したものであると仮定される。この配列は、インプットツリーのトポロジーを尊重した分岐プロセスに従って in silicoで進化する。
- 置換の割合mは入力木の枝の長さに基づいており、この割合はユーザーがリスケーリング係数で変更できる(例えば、リスケーリング係数0.5はすべての枝の長さを半分にする)。
- コード配列に作用する純化選択の効果を模倣するために、ユーザーが指定して、コドンの3つの位置で異なる置換率で配列を進化させることができる。
- いくつかの置換モデルを指定できる:JukeとCantor (JC69)、Kimura 2パラメータ(K2P)、Kimura 3パラメータ(K3P)、General Time Reversible (GTR)などの置換モデルを指定することができ、この場合、置換遷移/変換比κやその他のパラメータを指定する必要がある。
- CoreSimulでの相同組み換えは、シミュレーションデータセットに存在する相同な配列に置き換わることであり、水平遺伝子移動はシミュレーションゲノムの外部にある新しい配列を獲得することである。
- より現実的な条件を模倣するため、任意の時間に存在する異なる配列が同時に進化し、時間的に重なる配列のみが互いに組み換わることを許されている(祖先配列との組み換えは許されない)。各組み換えイベントのドナー配列は、時間セグメント内の配列セットからランダムに引き抜かれる。各組み換えイベントの位置は配列に沿ってランダムに選ばれ、その大きさはユーザーが指定した平均δの幾何分布によって定義される。
- CoreSimulはさらに、配列の分岐の関数として、ゲノムに沿った相同組み換えの不均一な割合をシミュレートできる。複数の研究により、相同組み換えの頻度は、対数線形関係に従って配列の分岐とともに減少することが実験的に決定されている[30,31,32,33,34,35,36]。その結果、配列の同一性が高い相同配列は、分岐の多い配列よりも組換えの可能性が高くなることが予測される。ユーザーが指定すると、CoreSimulはp = 10-πΦの関係を用いて相同組み換えの偏った確率を導入し、pは組換えの確率、πは配列の分岐度、Φは配列の分岐度と相同組換えの頻度の関係の傾きとする[33]。係数Φは種特異的であり、いくつかの種についてしか決定されていない[33]。従って、CoreSimulのデフォルトでは、Streptococcus pneumoniaeで実験的に推定されたΦ=18.1のパラメータが使用されており、枯草菌や大腸菌のようなΦの値が知られている他の種と比較すると中間的である[33]。
- 遺伝子の獲得と喪失の割合を指定することで、アクセサリー遺伝子の進化をシミュレートできる。CoreSimulのフレームワークでは、遺伝子の獲得は外部からの水平遺伝子移動としてモデル化される。遺伝子のgainとlossの速度は、ポアソン分布に従う置換率の関数として指定される(すなわち、枝の長さに比例する)。遺伝子の増減を指定しない場合、CoreSimulはコアゲノムの進化のみをシミュレートすることに注意。
引用
CoreSimul: a forward-in-time simulator of genome evolution for prokaryotes modeling homologous recombination
Louis-Marie Bobay
BMC Bioinformatics. 2020 Jun 24;21(1):264
関連
・