2019 3/12 タイトル修正
2019 3/12 コマンド追記、誤ったコメント削除
2020 1/19 GPU版のリンク追記
2020 5/4 3.6ツイート追記
2021 1/8 helpのバージョン更新、リンク切れ修正
GPU版
2020 3/13 構成を微修正、タイトル変更
20200 7/15
guppy v4.0.11
I got back into some @nanopore sequencing data I had from 2018 and ran basecalling again. Who knew I'd actually see the improved accuracy by eye 😮. At this pace, it will look like @illumina reads in another 2 years! pic.twitter.com/FfIrYQnUAz
— François Kroll (@francois_kroll) 2020年7月14日
2020 5/4
Guppy 3.6.0 is a marked improvement for PromethION @nanopore R9.4.1 data!!! Looking forward to the new announcements at LC2020!!! @NanoporeConf @BenedictPaten @kishwarshafin @HOlsenUCSC pic.twitter.com/wofInG39Be
— Miten Jain (@mitenjain) 2020年5月4日
Next up in the random nanopore read comparison: Bonito 0.1.5 vs Guppy 3.6.0: Bonito (left) seems to have a slightly higher accuracy due to fewer inserton/delitions but guppy maps a few more bases at the end of the read. pic.twitter.com/3tAEUZ3tYF
— Ola Wallerman (@OWallerman) 2020年5月2日
2021 /8現在v4.4.1が最新。
GuppyはOxford Nanoporeによって提供されているコマンドラインのbasecaller。 そしてポアを通過するDNAまたはRNAをbasecallingするために最新のリカレントニューラルネットワークアルゴリズムを利用してナノポアからのシグナルデータを解釈する。GPU GuppyはOxford Nanopore Technologiesのソフトウェア製品に安定した機能を実装しており、完全にサポートされている。 .fast5ファイルを入力として受け取り、ベースコール情報を付加した.fast5ファイル、処理された.fast5ファイル(1D^2は2回ランする)、fastqファイルを生成できる。ここではCPU版を使う流れを説明する。
マニュアルリンク
Log in - Oxford Nanopore Technologies

log inの必要あり。
インストール
mac10.12でバイナリをダウンロードしてテストした。
ハードウエア要件
- 4 GBのRAMと1 Dベースコールのスレッドあたり1 GB
- 4 GBのRAMと1 D 2ベースコールのスレッドあたり2 GB
- .debまたは.msiインストーラーの管理者アクセス
- インストール用に最大100 MBのドライブスペース、ベースコールされたリードファイル用に最低512 GBのストレージスペース(1 TB推奨)
- 外部GPU(GPU版)
Albacoreと同様、ダウンロードはOxford nanopore HPのsoftware downloadsから行う。
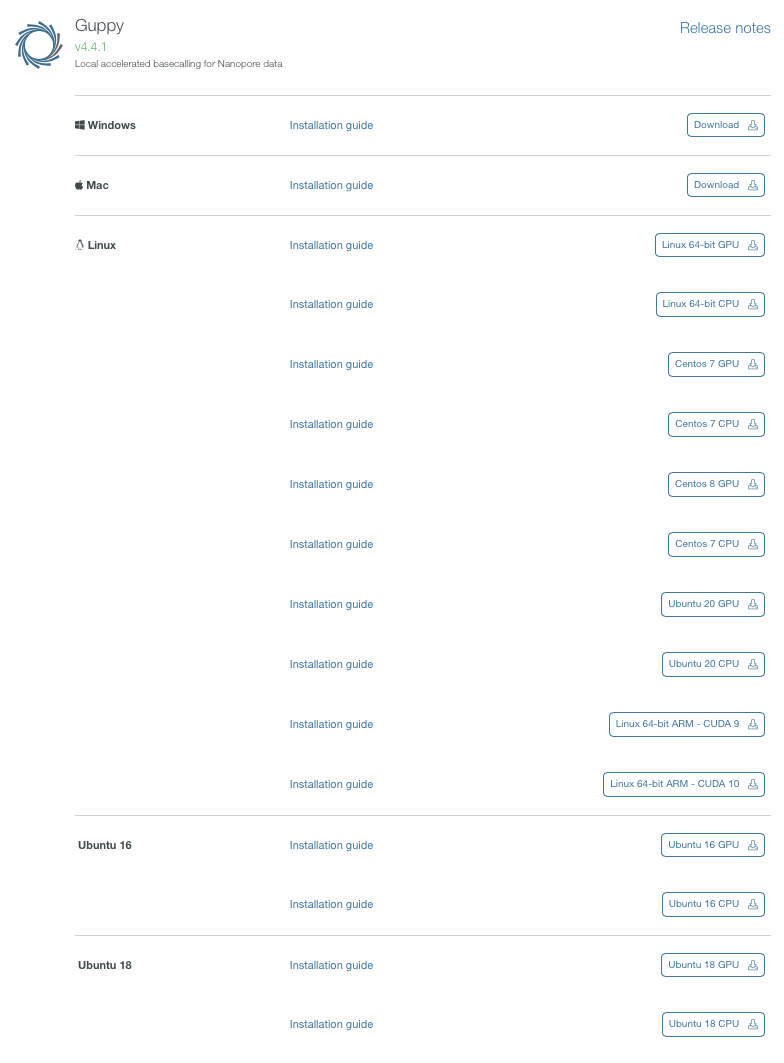
バイナリがダウンロードできる。ここではlinux 64bit向けをダウンロードした。gpu版のバイナリはcentos7版のみ用意されている。 その後、他のプラットフォームも公開されるようになった。ARM版もある。
cd ont-guppy-cpu/bin/
> ls -l
$ ls -l
total 18360
-rw-r--r--@ 1 user staff 196187 12 11 15:37 Nanopore Community TCs 09 September 2016.pdf
-rw-r--r--@ 1 user staff 57178 1 28 17:16 THIRD_PARTY_LICENSES
-rwxr-xr-x@ 1 user staff 950944 2 26 14:22 guppy_aligner
-rwxr-xr-x@ 1 user staff 1098576 2 26 14:22 guppy_barcoder
-rwxr-xr-x@ 1 user staff 1774404 2 26 14:22 guppy_basecall_server
-rwxr-xr-x@ 1 user staff 2707612 2 26 14:22 guppy_basecaller
-rwxr-xr-x@ 1 user staff 2598784 2 26 14:22 guppy_basecaller_1d2
> ./guppy_basecaller (v4.4.1)
: Guppy Basecalling Software, (C) Oxford Nanopore Technologies, Limited. Version 4.4.1+1c81d62
Usage:
With config file:"
guppy_basecaller -i <input path> -s <save path> -c <config file> [options]
With flowcell and kit name:
guppy_basecaller -i <input path> -s <save path> --flowcell <flowcell name>
--kit <kit name>
List supported flowcells and kits:
guppy_basecaller --print_workflows
Command line parameters:
--trim_threshold arg Threshold above which data will be trimmed
(in standard deviations of current level
distribution).
--trim_min_events arg Adapter trimmer minimum stride intervals
after stall that must be seen.
--max_search_len arg Maximum number of samples to search through
for the stall
--override_scaling Manually provide scaling parameters rather
than estimating them from each read.
--scaling_med arg Median current value to use for manual
scaling.
--scaling_mad arg Median absolute deviation to use for manual
scaling.
--trim_strategy arg Trimming strategy to apply: 'dna' or 'rna'
(or 'none' to disable trimming)
--dmean_win_size arg Window size for coarse stall event
detection
--dmean_threshold arg Threshold for coarse stall event detection
--jump_threshold arg Threshold level for rna stall detection
--pt_scaling Enable polyT/adapter max detection for read
scaling.
--pt_median_offset arg Set polyT median offset for setting read
scaling median (default 2.5)
--adapter_pt_range_scale arg Set polyT/adapter range scale for setting
read scaling median absolute deviation
(default 5.2)
--pt_required_adapter_drop arg Set minimum required current drop from
adapter max to polyT detection. (default
30.0)
--pt_minimum_read_start_index arg Set minimum index for read start sample
required to attempt polyT scaling. (default
30)
--as_model_file arg Path to JSON model file for adapter
scaling.
--as_gpu_runners_per_device arg Number of runners per GPU device for
adapter scaling.
--as_cpu_threads_per_scaler arg Number of CPU worker threads per adapter
scaler
--as_reads_per_runner arg Maximum reads per runner for adapter
scaling.
--as_num_scalers arg Number of parallel scalers for adapter
scaling.
-m [ --model_file ] arg Path to JSON model file.
-k [ --kernel_path ] arg Path to GPU kernel files location (only
needed if builtin_scripts is false).
-x [ --device ] arg Specify basecalling device: 'auto', or
'cuda:<device_id>'.
--builtin_scripts arg Whether to use GPU kernels that were
included at compile-time.
--chunk_size arg Stride intervals per chunk.
--chunks_per_runner arg Maximum chunks per runner.
--chunks_per_caller arg Soft limit on number of chunks in each
caller's queue. New reads will not be
queued while this is exceeded.
--high_priority_threshold arg Number of high priority chunks to process
for each medium priority chunk.
--medium_priority_threshold arg Number of medium priority chunks to process
for each low priority chunk.
--overlap arg Overlap between chunks (in stride
intervals).
--gpu_runners_per_device arg Number of runners per GPU device.
--cpu_threads_per_caller arg Number of CPU worker threads per
basecaller.
--num_callers arg Number of parallel basecallers to create.
--post_out Return full posterior matrix in output
fast5 file and/or called read message from
server.
--stay_penalty arg Scaling factor to apply to stay probability
calculation during transducer decode.
--qscore_offset arg Qscore calibration offset.
--qscore_scale arg Qscore calibration scale factor.
--temp_weight arg Temperature adjustment for weight matrix in
softmax layer of RNN.
--temp_bias arg Temperature adjustment for bias vector in
softmax layer of RNN.
--beam_cut arg Beam score cutoff for beam search decoding.
--beam_width arg Beam score cutoff for beam search decoding.
--qscore_filtering Enable filtering of reads into PASS/FAIL
folders based on min qscore.
--min_qscore arg Minimum acceptable qscore for a read to be
filtered into the PASS folder
--reverse_sequence arg Reverse the called sequence (for RNA
sequencing).
--u_substitution arg Substitute 'U' for 'T' in the called
sequence (for RNA sequencing).
--log_speed_frequency arg How often to print out basecalling speed.
--barcode_kits arg Space separated list of barcoding kit(s) or
expansion kit(s) to detect against. Must be
in double quotes.
--trim_barcodes Trim the barcodes from the output sequences
in the FastQ files.
--num_extra_bases_trim arg How vigorous to be in trimming the barcode.
Default is 0 i.e. the length of the
detected barcode. A positive integer means
extra bases will be trimmed, a negative
number is how many fewer bases (less
vigorous) will be trimmed.
--arrangements_files arg Files containing arrangements.
--lamp_arrangements_files arg Files containing lamp arrangements.
--score_matrix_filename arg File containing mismatch score matrix.
--start_gap1 arg Gap penalty for aligning before the
reference.
--end_gap1 arg Gap penalty for aligning after the
reference.
--open_gap1 arg Penalty for opening a new gap in the
reference.
--extend_gap1 arg Penalty for extending a gap in the
reference.
--start_gap2 arg Gap penalty for aligning before the query.
--end_gap2 arg Gap penalty for aligning after the query.
--open_gap2 arg Penalty for opening a new gap in the query.
--extend_gap2 arg Penalty for extending a gap in the query.
--min_score arg Minimum score to consider a valid
alignment.
--min_score_rear_override arg Minimum score to consider a valid alignment
for the rear barcode only (and min_score
will then be used for the front only when
this is set).
--min_score_mask arg Minimum score for a barcode context to
consider a valid alignment.
--front_window_size arg Window size for the beginning barcode.
--rear_window_size arg Window size for the ending barcode.
--require_barcodes_both_ends Reads will only be classified if there is a
barcode above the min_score at both ends of
the read.
--allow_inferior_barcodes Reads will still be classified even if both
the barcodes at the front and rear (if
applicable) were not the best scoring
barcodes above the min_score.
--detect_mid_strand_barcodes Search for barcodes through the entire
length of the read.
--min_score_mid_barcodes arg Minimum score for a barcode to be detected
in the middle of a read.
--lamp_kit arg LAMP barcoding kit to perform LAMP
detection against.
--min_score_lamp arg Minimum score for a LAMP barcode to be
classified.
--min_score_lamp_mask arg Minimum score for a LAMP barcode mask
context to be classified.
--min_score_lamp_target arg Minimum score for a LAMP target to be
classified.
--additional_context_bases arg Number of bases from a lamp FIP barcode
context to append to the front and rear of
the FIP barcode before performing matching.
Default is 2.
--min_length_lamp_context arg Minimum align length for a LAMP barcode
mask context to be classified.
--min_length_lamp_target arg Minimum align length for a LAMP target to
be classified.
--num_barcoding_buffers arg Number of GPU memory buffers to allocate to
perform barcoding into. Controls level of
parallelism on GPU for barcoding.
--num_mid_barcoding_buffers arg Number of GPU memory buffers to allocate to
perform barcoding into. Controls level of
parallelism on GPU for mid barcoding.
--num_barcode_threads arg Number of worker threads to use for
barcoding.
--calib_detect Enable calibration strand detection and
filtering.
--calib_reference arg Reference FASTA file containing calibration
strand.
--calib_min_sequence_length arg Minimum sequence length for reads to be
considered candidate calibration strands.
--calib_max_sequence_length arg Maximum sequence length for reads to be
considered candidate calibration strands.
--calib_min_coverage arg Minimum reference coverage to pass
calibration strand detection.
--print_workflows Output available workflows.
--flowcell arg Flowcell to find a configuration for
--kit arg Kit to find a configuration for
-a [ --align_ref ] arg Path to alignment reference.
--bed_file arg Path to .bed file containing areas of
interest in reference genome.
--num_alignment_threads arg Number of worker threads to use for
alignment.
-z [ --quiet ] Quiet mode. Nothing will be output to
STDOUT if this option is set.
--trace_categories_logs arg Enable trace logs - list of strings with
the desired names.
--verbose_logs Enable verbose logs.
--trace_domains_log arg List of trace domains to include in verbose
logging (if enabled), '*' for all.
--trace_domains_config arg Configuration file containing list of trace
domains to include in verbose logging (if
enabled), this will override
--trace_domain_logs
--disable_pings Disable the transmission of telemetry
pings.
--ping_url arg URL to send pings to
--ping_segment_duration arg Duration in minutes of each ping segment.
--progress_stats_frequency arg Frequency in seconds in which to report
progress statistics, if supplied will
replace the default progress display.
-q [ --records_per_fastq ] arg Maximum number of records per fastq file, 0
means use a single file (per worker, per
run id).
--read_batch_size arg Maximum batch size, in reads, for grouping
input files.
--compress_fastq Compress fastq output files with gzip.
-i [ --input_path ] arg Path to input fast5 files.
--input_file_list arg Optional file containing list of input
fast5 files to process from the input_path.
-s [ --save_path ] arg Path to save fastq files.
-l [ --read_id_list ] arg File containing list of read ids to filter
to
-r [ --recursive ] Search for input files recursively.
--fast5_out Choice of whether to do fast5 output.
--bam_out Choice of whether to do BAM file output.
--bam_methylation_threshold arg The value below which a predicted
methylation probability will not be emitted
into a BAM file, expressed as a percentage.
Default is 5.0(%).
--resume Resume a previous basecall run using the
same output folder.
--client_id arg Optional unique identifier (non-negative
integer) for this instance of the Guppy
Client Basecaller, if supplied will form
part of the output filenames.
--nested_output_folder If flagged output fastq files will be
written to a nested folder structure, based
on: protocol_group/sample/protocol/qscore_p
ass_fail/barcode_arrangement/
--max_queued_reads arg Maximum number of reads to be submitted for
processing at any one time.
-h [ --help ] produce help message
-v [ --version ] print version number
-c [ --config ] arg Config file to use
-d [ --data_path ] arg Path to use for loading any data files the
application requires.
> ./guppy_basecaller_1d2 (Version 2.3.5)
$ ./guppy_basecaller_1d2
: Guppy 1D-Squared Basecalling Software, (C) Oxford Nanopore Technologies, Limited. Version 2.3.5+53a111f6
Command line parameters:
--print_workflows Output available workflows.
--flowcell arg Flowcell to find a configuration for
--kit arg Kit to find a configuration for
-m [ --model_file ] arg Path to JSON model file.
--chunk_size arg Stride intervals per chunk.
--chunks_per_runner arg Maximum chunks per runner.
--chunks_per_caller arg Soft limit on number of chunks in each
caller's queue. New reads will not be queued
while this is exceeded.
--overlap arg Overlap between chunks (in stride intervals).
--gpu_runners_per_device arg Number of runners per GPU device.
--cpu_threads_per_caller arg Number of CPU worker threads per basecaller.
--num_callers arg Number of parallel basecallers to create.
--stay_penalty arg Scaling factor to apply to stay probability
calculation during transducer decode.
--qscore_offset arg Qscore calibration offset.
--qscore_scale arg Qscore calibration scale factor.
--temp_weight arg Temperature adjustment for weight matrix in
softmax layer of RNN.
--temp_bias arg Temperature adjustment for bias vector in
softmax layer of RNN.
--hp_correct arg Whether to use homopolymer correction during
decoding.
--builtin_scripts arg Whether to use GPU kernels that were included
at compile-time.
-x [ --device ] arg Specify basecalling device: 'auto', or
'cuda:<device_id>'.
-k [ --kernel_path ] arg Path to GPU kernel files location (only
needed if builtin_scripts is false).
-z [ --quiet ] Quiet mode. Nothing will be output to STDOUT
if this option is set.
--trace_categories_logs arg Enable trace logs - list of strings with the
desired names.
--verbose_logs Enable verbose logs.
--qscore_filtering Enable filtering of reads into PASS/FAIL
folders based on min qscore.
--min_qscore arg Minimum acceptable qscore for a read to be
filtered into the PASS folder
--disable_pings Disable the transmission of telemetry pings.
--ping_url arg URL to send pings to
--ping_segment_duration arg Duration in minutes of each ping segment.
--calib_detect Enable calibration strand detection and
filtering.
--calib_reference arg Reference FASTA file containing calibration
strand.
--calib_min_sequence_length arg Minimum sequence length for reads to be
considered candidate calibration strands.
--calib_max_sequence_length arg Maximum sequence length for reads to be
considered candidate calibration strands.
--calib_min_coverage arg Minimum reference coverage to pass
calibration strand detection.
--score_matrix arg Path to mismatch matrix for prior label
alignment
-q [ --records_per_fastq ] arg Maximum number of records per fastq file (0
means use a single file).
--winsize1 arg Short window length for event detection.
--winsize2 arg Long window length for event detection.
--threshold1 arg Shirt time-scale threshold for event
detection.
--threshold2 arg Long time-scale threshold for event
detection.
--band_size arg Band size for 1d-squared alignment table.
--pa_band_size arg Band size for prior-alignment table.
--gap_penalty arg Gap penalty for prior label alignment.
--start_end_penalty arg Overhang penalty for prior label alignment.
--reverse_sequence arg Reverse the called sequence (for RNA
sequencing).
--u_substitution arg Substute 'U' for 'T' in the called sequence
(for RNA sequencing).
-i [ --input_path ] arg Path to input fast5 files.
-f [ --index_file ] arg Index file from 1D basecall.
-s [ --save_path ] arg Path to save fastq files.
-p [ --port ] arg Hostname and port for connecting to basecall
service (ie 'myserver:5555'), or port only
(ie '5555'), in which case localhost is
assumed.
-r [ --recursive ] Search for input files recursively.
--override_scaling Manually provide scaling parameters rather
than estimating them from each read.
--scaling_med arg Median current value to use for manual
scaling.
--scaling_mad arg Median absolute deviation to use for manual
scaling.
-h [ --help ] produce help message
-v [ --version ] print version number
-c [ --config ] arg Config file to use
-d [ --data_path ] arg Path to use for loading any data files the
application requires.
> ./guppy_barcoder
$ ./guppy_barcoder
Usage:
guppy_barcoder -i <input fastq path> -s <save path>
With kit name:
guppy_barcoder -i <input fastq path> -s <save path> --barcode_kits <kit name>
--kit <kit name>
List supported barcoding kits:
guppy_barcoder --print_kits
Command line parameters:
-z [ --quiet ] Quiet mode. Nothing will be output to stdout
if this option is set.
-t [ --worker_threads ] arg Number of worker threads.
-i [ --input_path ] arg Path to input fastq files.
-s [ --save_path ] arg Path to save fastq files.
-r [ --recursive ] Search for input file recursively.
--trace_categories_logs arg Enable trace logs - list of strings with the
desired names.
--verbose_logs Enable verbose logs.
--print_kits Output all available barcoding kits.
--barcode_kits arg Space separated list of barcoding kit(s) or
expansion kit(s) to detect against. Must be in
double quotes.
-q [ --records_per_fastq ] arg Maximum number of records per fastq file, 0
means use a single file (per run id).
--arrangements_files arg Files containing arrangements.
--score_matrix_filename arg File containing mismatch score matrix.
--start_gap1 arg Gap penalty for aligning before the reference.
--end_gap1 arg Gap penalty for aligning after the reference.
--open_gap1 arg Penalty for opening a new gap in the
reference.
--extend_gap1 arg Penalty for extending a gap in the reference.
--start_gap2 arg Gap penalty for aligning before the query.
--end_gap2 arg Gap penalty for aligning after the query.
--open_gap2 arg Penalty for opening a new gap in the query.
--extend_gap2 arg Penalty for extending a gap in the query.
--min_score arg Minimum score to consider a valid alignment.
--front_window_size arg Window size for the beginning barcode.
--rear_window_size arg Window size for the ending barcode.
-h [ --help ] produce help message
-v [ --version ] print version number
-c [ --config ] arg Config file to use
-d [ --data_path ] arg Path to use for loading any data files the
application requires.
> ./guppy_basecall_server
$ ./guppy_basecall_server
: Guppy Basecall Service Software, (C) Oxford Nanopore Technologies, Limited. Version 2.3.5+53a111f6
Usage:
With config file:
guppy_basecall_server -c <config file> --port <server listen port>
--log_path <log file path> [options]
With flowcell and kit:
guppy_basecall_server --flowcell <flowcell name> --kit <kit name>
--port <server listen port> --log_path <log file path> [options]
List supported flowcells and kits:
guppy_basecall_server --print_workflows
Command line parameters:
--print_workflows Output available workflows.
--flowcell arg Flowcell to find a configuration for
--kit arg Kit to find a configuration for
-m [ --model_file ] arg Path to JSON model file.
--chunk_size arg Stride intervals per chunk.
--chunks_per_runner arg Maximum chunks per runner.
--chunks_per_caller arg Soft limit on number of chunks in each
caller's queue. New reads will not be queued
while this is exceeded.
--overlap arg Overlap between chunks (in stride intervals).
--gpu_runners_per_device arg Number of runners per GPU device.
--cpu_threads_per_caller arg Number of CPU worker threads per basecaller.
--num_callers arg Number of parallel basecallers to create.
--stay_penalty arg Scaling factor to apply to stay probability
calculation during transducer decode.
--qscore_offset arg Qscore calibration offset.
--qscore_scale arg Qscore calibration scale factor.
--temp_weight arg Temperature adjustment for weight matrix in
softmax layer of RNN.
--temp_bias arg Temperature adjustment for bias vector in
softmax layer of RNN.
--hp_correct arg Whether to use homopolymer correction during
decoding.
--builtin_scripts arg Whether to use GPU kernels that were included
at compile-time.
-x [ --device ] arg Specify basecalling device: 'auto', or
'cuda:<device_id>'.
-k [ --kernel_path ] arg Path to GPU kernel files location (only
needed if builtin_scripts is false).
-z [ --quiet ] Quiet mode. Nothing will be output to STDOUT
if this option is set.
--trace_categories_logs arg Enable trace logs - list of strings with the
desired names.
--verbose_logs Enable verbose logs.
--qscore_filtering Enable filtering of reads into PASS/FAIL
folders based on min qscore.
--min_qscore arg Minimum acceptable qscore for a read to be
filtered into the PASS folder
--disable_pings Disable the transmission of telemetry pings.
--ping_url arg URL to send pings to
--ping_segment_duration arg Duration in minutes of each ping segment.
--calib_detect Enable calibration strand detection and
filtering.
--calib_reference arg Reference FASTA file containing calibration
strand.
--calib_min_sequence_length arg Minimum sequence length for reads to be
considered candidate calibration strands.
--calib_max_sequence_length arg Maximum sequence length for reads to be
considered candidate calibration strands.
--calib_min_coverage arg Minimum reference coverage to pass
calibration strand detection.
--ipc_threads arg Number of threads to use for inter-process
communication.
--max_queued_reads arg Maximum number of reads in input queue.
-l [ --log_path ] arg Path to save log file.
-p [ --port ] arg Port for hosting service. Specify "auto" to
make server automatically search for a free
port.
-h [ --help ] produce help message
-v [ --version ] print version number
-c [ --config ] arg Config file to use
-d [ --data_path ] arg Path to use for loading any data files the
application requires.
> ./guppy_aligner
$ ./guppy_aligner
Usage:
guppy_aligner -i <input fastq path> -s <output SAM path>
--align_ref <reference file>
Command line parameters:
-z [ --quiet ] Quiet mode. Nothing will be output to stdout if
this option is set.
-t [ --worker_threads ] arg Number of worker threads.
-i [ --input_path ] arg Path to input fastq files.
-s [ --save_path ] arg Path to save fastq files.
-r [ --recursive ] Search for input file recursively.
--trace_categories_logs arg Enable trace logs - list of strings with the
desired names.
--verbose_logs Enable verbose logs.
-a [ --align_ref ] arg Path to alignment reference.
--min_coverage arg Minimum coverage to accept an alignment.
-h [ --help ] produce help message
-v [ --version ] print version number
-c [ --config ] arg Config file to use
-d [ --data_path ] arg Path to use for loading any data files the
application requires.
対応フローセルとキット
> guppy_basecaller --print_workflows
$ guppy_basecaller --print_workflows
Available flowcell + kit combinations are:
flowcell kit barcoding config_name
FLO-MIN106 SQK-DCS108 dna_r9.4.1_450bps
FLO-MIN106 SQK-DCS109 dna_r9.4.1_450bps
FLO-MIN106 SQK-LRK001 dna_r9.4.1_450bps
FLO-MIN106 SQK-LSK108 dna_r9.4.1_450bps
FLO-MIN106 SQK-LSK109 dna_r9.4.1_450bps
FLO-MIN106 SQK-LWP001 dna_r9.4.1_450bps
FLO-MIN106 SQK-PCS108 dna_r9.4.1_450bps
FLO-MIN106 SQK-PCS109 dna_r9.4.1_450bps
FLO-MIN106 SQK-PSK004 dna_r9.4.1_450bps
FLO-MIN106 SQK-RAD002 dna_r9.4.1_450bps
FLO-MIN106 SQK-RAD003 dna_r9.4.1_450bps
FLO-MIN106 SQK-RAD004 dna_r9.4.1_450bps
FLO-MIN106 SQK-RAS201 dna_r9.4.1_450bps
FLO-MIN106 SQK-RLI001 dna_r9.4.1_450bps
FLO-MIN106 VSK-VBK001 dna_r9.4.1_450bps
FLO-MIN106 VSK-VSK001 dna_r9.4.1_450bps
FLO-MIN106 SQK-RBK001 included dna_r9.4.1_450bps
FLO-MIN106 SQK-RBK004 included dna_r9.4.1_450bps
FLO-MIN106 SQK-RLB001 included dna_r9.4.1_450bps
FLO-MIN106 SQK-LWB001 included dna_r9.4.1_450bps
FLO-MIN106 SQK-PBK004 included dna_r9.4.1_450bps
FLO-MIN106 SQK-RAB201 included dna_r9.4.1_450bps
FLO-MIN106 SQK-RAB204 included dna_r9.4.1_450bps
FLO-MIN106 SQK-RPB004 included dna_r9.4.1_450bps
FLO-MIN106 VSK-VMK001 included dna_r9.4.1_450bps
FLO-PRO001 SQK-LSK109 dna_r9.4.1_450bps_prom
FLO-PRO001 SQK-DCS109 dna_r9.4.1_450bps_prom
FLO-PRO001 SQK-PCS109 dna_r9.4.1_450bps_prom
FLO-PRO002 SQK-LSK109 dna_r9.4.1_450bps_prom
FLO-PRO002 SQK-DCS109 dna_r9.4.1_450bps_prom
FLO-PRO002 SQK-PCS109 dna_r9.4.1_450bps_prom
FLO-MIN107 SQK-DCS108 dna_r9.5_450bps
FLO-MIN107 SQK-DCS109 dna_r9.5_450bps
FLO-MIN107 SQK-LRK001 dna_r9.5_450bps
FLO-MIN107 SQK-LSK108 dna_r9.5_450bps
FLO-MIN107 SQK-LSK109 dna_r9.5_450bps
FLO-MIN107 SQK-LSK308 dna_r9.5_450bps
FLO-MIN107 SQK-LSK309 dna_r9.5_450bps
FLO-MIN107 SQK-LSK319 dna_r9.5_450bps
FLO-MIN107 SQK-LWP001 dna_r9.5_450bps
FLO-MIN107 SQK-PCS108 dna_r9.5_450bps
FLO-MIN107 SQK-PCS109 dna_r9.5_450bps
FLO-MIN107 SQK-PSK004 dna_r9.5_450bps
FLO-MIN107 SQK-RAD002 dna_r9.5_450bps
FLO-MIN107 SQK-RAD003 dna_r9.5_450bps
FLO-MIN107 SQK-RAD004 dna_r9.5_450bps
FLO-MIN107 SQK-RAS201 dna_r9.5_450bps
FLO-MIN107 SQK-RLI001 dna_r9.5_450bps
FLO-MIN107 VSK-VBK001 dna_r9.5_450bps
FLO-MIN107 VSK-VSK001 dna_r9.5_450bps
FLO-MIN107 SQK-LWB001 included dna_r9.5_450bps
FLO-MIN107 SQK-PBK004 included dna_r9.5_450bps
FLO-MIN107 SQK-RAB201 included dna_r9.5_450bps
FLO-MIN107 SQK-RAB204 included dna_r9.5_450bps
FLO-MIN107 SQK-RBK001 included dna_r9.5_450bps
FLO-MIN107 SQK-RBK004 included dna_r9.5_450bps
FLO-MIN107 SQK-RLB001 included dna_r9.5_450bps
FLO-MIN107 SQK-RPB004 included dna_r9.5_450bps
FLO-MIN107 VSK-VMK001 included dna_r9.5_450bps
FLO-MIN106 SQK-RNA001 rna_r9.4.1_70bps
FLO-MIN106 SQK-RNA002 rna_r9.4.1_70bps
FLO-MIN107 SQK-RNA001 rna_r9.4.1_70bps
FLO-MIN107 SQK-RNA002 rna_r9.4.1_70bps
FLO-PRO001 SQK-RNA002 rna_r9.4.1_70bps_prom
FLO-PRO002 SQK-RNA002 rna_r9.4.1_70bps_prom
実行方法
フローセル、kit名、入出力(*1)等を指定して実行する。-r (--recursive)をつけるとサブディレクトリも含めてbasecallingされる(*1)。
guppy_basecaller --flowcell FLO-MIN106 --kit SQK-LSK109 \
--cpu_threads_per_caller 4 --num_callers 4 \
-i fast5/input_dir -s output_dir -r
出力(サブディレクトリ1つ分)

1D^2のbase callingの場合、--fast5_outフラグを立てて上のコマンドを実行し、得られたfast5ファイルをguppy_basecaller_1d2で再びbase callingする2stepで行う(未テスト)。
引用
nanoporetech Guppy 2.3.5
https://community.nanoporetech.com/protocols/Guppy-protocol-preRev/v/gpb_2003_v1_revh_14dec2018
参考
*1
ツイッターであかまるさんに教えていただきました。