Viromicsは毎年数百万個のウイルスゲノムと断片を産生し、従来の配列比較法を圧倒している。Vclustは、Lempel-Ziv構文解析によって平均塩基同一性を決定し、権威ある viral genomics and taxonomy consortiaによって承認された閾値でウイルスゲノムをクラスタリングする新しいアプローチである。Vclustは既存のツールと比較して優れた精度と効率を示し、ミッドレンジのワークステーションで数百万個のウイルスゲノムを数時間でクラスタリングした。
webサーバー
使うにはローカルのfastaファイルを指定する。

インストール
- https://github.com/refresh-bio/kmer-db
- https://github.com/refresh-bio/LZ-ANI(紹介)
- https://github.com/refresh-bio/clusty
git clone --recurse-submodules https://github.com/refresh-bio/vclust
cd vclust && make -j
> ./vclust.py -h
usage: vclust.py [-v] [-h] {prefilter,align,cluster,info} ...
vclust.py v.1.2.7: calculate ANI and cluster virus (meta)genome sequences
positional arguments:
{prefilter,align,cluster,info}
prefilter Prefilter genome pairs for alignment
align Align genome sequences and calculate ANI metrics
cluster Cluster genomes based on ANI thresholds
info Show information about the tool and its dependencies
options:
-v, --version Display the tool's version and exit
-h, --help Show this help message and exit
> ./vclust.py prefilter -h
usage: vclust.py prefilter -i <file> -o <file> [-k <int>] [--min-kmers <int>] [--min-ident <float>] [--batch-size <int>] [--kmers-fraction <float>] [--max-seqs <int>] [--keep_temp] [--bin <file>] [--bin-fasta <file>] [-t <int>] [-v] [-h]
required arguments:
-i, --in <file> Input FASTA file or directory with FASTA files
-o, --out <file> Output filename
options:
-k, --k <int> Size of k-mer for Kmer-db [25]
--min-kmers <int> Filter genome pairs based on minimum number of shared k-mers [10]
--min-ident <float> Filter genome pairs based on minimum sequence identity of the shorter sequence (0-1) [0.7]
--batch-size <int> Process a multifasta file in smaller batches of n FASTA sequences. This option reduces memory at the expense of speed. By default, no batch [0]
--kmers-fraction <float> Fraction of k-mers to analyze for each genome (0-1). A lower value reduces RAM usage and speeds up processing (affects sensitivity) [1.0]
--max-seqs <int> Maximum number of sequences allowed to pass the prefilter per query. Only the sequences with the highest identity to the query are reported. This option reduces RAM usage and speeds up processing (affects sensitivity). By default, all sequences that pass the prefilter are
reported [0]
--keep_temp Keep temporary Kmer-db files [False]
--bin <file> Path to the Kmer-db binary [/home/kazu/Documents/vclust/bin/kmer-db]
--bin-fasta <file> Path to the multi-fasta-split binary [/home/kazu/Documents/vclust/bin/multi-fasta-split]
-t, --threads <int> Number of threads (all by default) [64]
-v, --verbose Show Kmer-db progress
-h, --help Show this help message and exit
> ./vclust.py align -h
usage: vclust.py align -i <file> -o <file> [--filter <file>] [--filter-threshold <float>] [--outfmt <str>] [--out-aln <file>] [--out-ani <float>] [--out-tani <float>] [--out-gani <float>] [--out-qcov <float>] [--out-rcov <float>] [--bin <file>] [--mal <int>] [--msl <int>] [--mrd <int>] [--mqd <int>]
[--reg <int>] [--aw <int>] [--am <int>] [--ar <int>] [-t <int>] [-v] [-h]
required arguments:
-i, --in <file> Input FASTA file or directory with FASTA files
-o, --out <file> Output filename
options:
--filter <file> Path to filter file (output of prefilter)
--filter-threshold <float> Align genome pairs above the threshold (0-1) [0]
--outfmt <str> Output format [standard]
choices: lite,standard,complete
--out-aln <file> Write alignments to the specified tsv file (optional).
--out-ani <float> Min. ANI to output (0-1) [0]
--out-tani <float> Min. tANI to output (0-1) [0]
--out-gani <float> Min. gANI to output (0-1) [0]
--out-qcov <float> Min. query coverage (aligned fraction) to output (0-1) [0]
--out-rcov <float> Min. reference coverage (aligned fraction) to output (0-1) [0]
--bin <file> Path to the LZ-ANI binary [/home/kazu/Documents/vclust/bin/lz-ani]
--mal <int> Min. anchor length [11]
--msl <int> Min. seed length [7]
--mrd <int> Max. dist. between approx. matches in reference [40]
--mqd <int> Max. dist. between approx. matches in query [40]
--reg <int> Min. considered region length [35]
--aw <int> Approx. window length [15]
--am <int> Max. no. of mismatches in approx. window [7]
--ar <int> Min. length of run ending approx. extension [3]
-t, --threads <int> Number of threads (all by default) [64]
-v, --verbose Show LZ-ANI progress
-h, --help Show this help message and exit
> ./vclust.py cluster -h
usage: vclust.py cluster -i <file> -o <file> --ids <file> [-r] [--algorithm <str>] [--metric <str>] [--tani <float>] [--gani <float>] [--ani <float>] [--qcov <float>] [--rcov <float>] [--len_ratio <float>] [--num_alns <int>] [--leiden-resolution <float>] [--leiden-beta <float>] [--leiden-iterations <int>]
[--bin <file>] [-v] [-h]
required arguments:
-i, --in <file> Input file with ANI metrics (tsv)
-o, --out <file> Output filename
--ids <file> Input file with sequence identifiers (tsv)
options:
-r, --out-repr Output a representative genome for each cluster instead of numerical cluster identifiers. The representative genome is selected as the one with the longest sequence. [False]
--algorithm <str> Clustering algorithm [single]
* single: Single-linkage (connected component)
* complete: Complete-linkage
* uclust: UCLUST
* cd-hit: Greedy incremental
* set-cover: Greedy set-cover (MMseqs2)
* leiden: Leiden algorithm
--metric <str> Similarity metric for clustering [tani]
choices: tani,gani,ani
--tani <float> Min. total ANI (0-1) [0]
--gani <float> Min. global ANI (0-1) [0]
--ani <float> Min. ANI (0-1) [0]
--qcov <float> Min. query coverage/aligned fraction (0-1) [0]
--rcov <float> Min. reference coverage/aligned fraction (0-1) [0]
--len_ratio <float> Min. length ratio between shorter and longer sequence (0-1) [0]
--num_alns <int> Max. number of local alignments between two genomes; 0 means all genome pairs are allowed. [0]
--leiden-resolution <float> Resolution parameter for the Leiden algorithm [0.7]
--leiden-beta <float> Beta parameter for the Leiden algorithm [0.01]
--leiden-iterations <int> Number of iterations for the Leiden algorithm [2]
--bin <file> Path to the Clusty binary [/home/kazu/Documents/vclust/bin/clusty]
-v, --verbose Show Clusty progress
-h, --help Show this help message and exit
> ./vclust.py info
Vclust 1.2.7
kmer-db ok
multi-fasta-split ok
lz-ani ok
clusty ok
テストラン
3つのステップ;プレフィルター、整列とANI計算、クラスタリング、のコマンドが用意されている。
1、vclust.py prefilter
ウィルスゲノムのmulti-fastaを指定する。prefilterサブコマンドでは類似ゲノム配列ペアはプレフィルタリングされる。
cd vclust
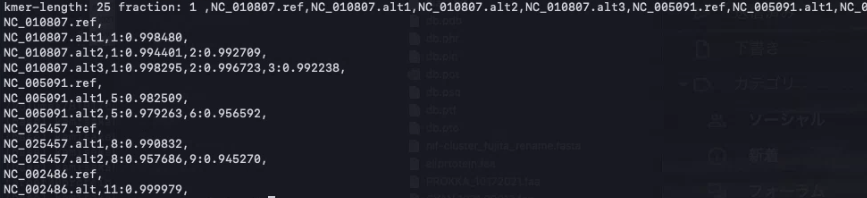
./vclust.py prefilter -i example/multifasta.fna -o fltr.txt
2、vclust.py align
類似ゲノム配列ペアを整列し、ペアワイズ ANIを計算する。1の出力ファイル”fltr.txt”を指定する。
./vclust.py align -i example/multifasta.fna -o ani.tsv --filter fltr.txt
- --filter Path to filter file (output of prefilter)
ani.tsvが出力される。
> column -t ani.tsv

3,vclust.py cluster
与えられたANI指標と最小閾値に基づいてゲノム配列をクラスタリングする。
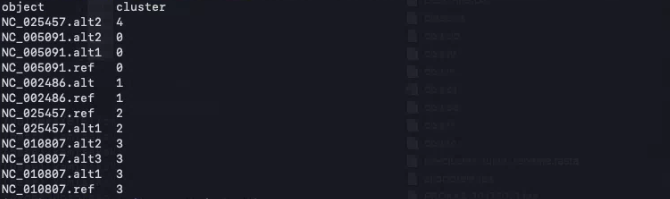
./vclust.py cluster -i ani.tsv -o clusters.tsv --ids ani.ids.tsv --metric ani --ani 0.95
- --metric Similarity metric for clustering [tani]
choices: tani, gani, ani
> column -t clusters.tsv
引用
Ultrafast and accurate sequence alignment and clustering of viral genomes
Andrzej Zielezinski, Adam Gudyś, Jakub Barylski, Krzysztof Siminski, Piotr Rozwalak, Bas E. Dutilh, Sebastian Deorowicz
bioRxiv, Posted July 02, 2024.