細菌ゲノムは、遺伝子含有量と配列変異の両方において異なっており、抗菌薬に対する感受性やワクチン誘発免疫の変異など、広範な表現型の多様性の根底にある。重要な変異を同定し定量化するためには、集団内のすべての遺伝子を予測し、機能アノテーションを行い、「パンゲノム 」としてクラスタリングする必要がある。ゲノムデータは大量にあるにもかかわらず、遺伝子の予測やアノテーションは個々のゲノムに対して個別に行われており、計算効率が悪く、ゲノム間で一貫性がないことが多い。ggCallerは、遺伝子予測、機能アノテーション、クラスタリングを、集団全体のde Bruijnグラフを用いた単一のワークフローに統合することで、遺伝子アノテーションの冗長性を除去し、より正確な遺伝子予測とオルソログクラスタリングを実現する。ggCallerはまた、細菌ゲノムワイド関連研究への重要な拡張であり、機能解析のためにアノテーションされたグラフのクエリを可能にする。本著者らは有意な関連を持つDNA配列を機能的にアノテーションすることで、このアプリケーションを強調している。
ggCallerのワークフロー ggCallerは2つのセクション: ORF同定(ステップ1-4)とORFクラスタリング+フィルタリング(ステップ5-8)に分けられる。(1) BifrostによりアセンブリからDBGを生成する。(2) すべての停止コドンを同定し、停止頻度を計算する(DBG中の停止コドンの総数/DBG中のコドンの総数)。(3a) 停止コドンを含む最初のノードから開始し、深さ優先探索(DFS)を用いて、開始ノード内のすべての停止コドンと、同じリーディングフレーム内の下流の停止コドンとを対にする。(3b) DFSの間、パスはFM-インデックスと比較され、誤ったパスを除去する。(4) ORFは、翻訳開始部位の配列、ゲノムカバレッジ(ノードで共有される色の数で与えられる)、およびこの開始コドンが他の潜在的なオルソログで選択される頻度に基づいてスコア化された開始コドンを同定することによって定義される。停止コドンを含むすべてのノードについて、ステップ3と4を繰り返す。(5) ORFはCOGにクラスタ化され、ノード共有を使って検索空間を縮小する。(6) Balrogを使用して、各COGの中心配列のみを使用して、1残基あたりの平均スコアを生成する。この1残基あたりの平均スコアは、中心配列の各クラスタ内の各ORFのスコアに使用される。(7) Bellman-Ford アルゴリズム(Bellman 1958; Ford and Fulkerson 1962)を用いて、DBG 内で重複する遺伝子について最高得点のタイリングパスを計算し、「真の」遺伝子コールセットを作成する。(8) 遺伝子コールとシンテニー情報を用いて遺伝子グラフを作成する。 Panarooの改良版を用いて、サポートが不十分な遺伝子コールを除去し、クラスターをアノテーションし、ミスした遺伝子/偽遺伝子をリコールする。
Documentation
https://ggcaller.readthedocs.io/en/latest/
Quickstart
https://ggcaller.readthedocs.io/en/latest/quickstart.html
Our work – ggCaller
https://www.bacpop.org/blog/ggcaller/
"ゲノム間で同じ遺伝子が同じように予測されるようにする方法が必要である。これを実現するために、ggCallerとしても知られるgraph-gene-callerを開発した。ggCallerは、ゲノムの遺伝子を1つずつ予測するのではなく、「グラフ」と呼ばれるネットワークのような構造の中で遺伝子をまとめて予測する。グラフをスキャンすることにより、ggCallerはゲノム間で共有される遺伝子を特定することができ、similarな遺伝子が共有されているゲノム間で予測されることを保証する。"
インストール
公開されているdocker image(著者らのもの)を使った。
git clone --recursive https://github.com/samhorsfield96/ggCaller
cd ggCaller
mamba env create -f environment_linux.yml
conda activate ggc_env
python setup.py install
#docker
docker pull samhorsfield96/ggcaller:latest
> ggcaller -h
usage: ggcaller [-h] [--graph GRAPH] [--colours COLOURS] [--not-ref] [--refs REFS] [--reads READS] [--query QUERY] [--codons CODONS] [--kmer KMER] [--save] [--data DATA] [--all-seq-in-graph] [--out OUT] [--max-path-length MAX_PATH_LENGTH] [--min-orf-length MIN_ORF_LENGTH]
[--score-tolerance SCORE_TOLERANCE] [--max-ORF-overlap MAX_ORF_OVERLAP] [--min-path-score MIN_PATH_SCORE] [--min-orf-score MIN_ORF_SCORE] [--max-orf-orf-distance MAX_ORF_ORF_DISTANCE] [--query-id QUERY_ID] [--no-filter] [--no-write-idx] [--no-write-graph]
[--repeat] [--no-clustering] [--no-refind] [--identity-cutoff IDENTITY_CUTOFF] [--len-diff-cutoff LEN_DIFF_CUTOFF] [--family-threshold FAMILY_THRESHOLD] [--merge-paralogs] [--clean-mode {strict,moderate,sensitive}]
[--annotation {none,fast,sensitive,ultrasensitive}] [--diamonddb ANNOTATION_DB] [--hmmdb HMM_DB] [--evalue EVALUE] [--truncation-threshold TRUNCATION_THRESHOLD] [--search-radius SEARCH_RADIUS] [--refind-prop-match REFIND_PROP_MATCH]
[--min-trailing-support MIN_TRAILING_SUPPORT] [--trailing-recursive TRAILING_RECURSIVE] [--edge-support-threshold EDGE_SUPPORT_THRESHOLD] [--length-outlier-support-proportion LENGTH_OUTLIER_SUPPORT_PROPORTION] [--min-edge-support-sv MIN_EDGE_SUPPORT_SV]
[--no-clean-edges] [--alignment {core,pan}] [--aligner {def,ref}] [--core-threshold CORE] [--no-variants] [--ignore-pseduogenes] [--balrog-db BALROG_DB] [--quiet] [--threads THREADS] [--version]
Generates ORFs from a Bifrost graph.
optional arguments:
-h, --help show this help message and exit
Input/Output options:
--graph GRAPH Bifrost GFA file generated by Bifrost build.
--colours COLOURS Bifrost colours file generated by Bifrost build.
--not-ref If using existing graph, was not graph built exclusively with assembled genomes. [Default = False]
--refs REFS List of reference genomes (one file path per line).
--reads READS List of read files (one file path per line).
--query QUERY List of unitig sequences to query (either FASTA or one sequence per line)
--codons CODONS JSON file containing start and stop codon sequences.
--kmer KMER K-mer size used in Bifrost build (bp). [Default = 31]
--save Save graph objects for sequence querying. [Default = False]
--data DATA Directory containing data from previous ggCaller run generated via "--save"
--all-seq-in-graph Retains all DNA sequence for each gene cluster in the Panaroo graph output. Off by default as it uses a large amount of space.
--out OUT Output directory
ggCaller traversal and gene-calling cut-off settings:
--max-path-length MAX_PATH_LENGTH
Maximum path length during ORF finding (bp). [Default = 20000]
--min-orf-length MIN_ORF_LENGTH
Minimum ORF length to return (bp). [Default = 90]
--score-tolerance SCORE_TOLERANCE
Length probability tolerance for shorter alternative start sites. If within tolerance,ggCaller will check if start coverage and BALROG score are both higher in shorter ORF. [Default = 0.2]
--max-ORF-overlap MAX_ORF_OVERLAP
Maximum overlap allowed between overlapping ORFs. [Default = 60]
--min-path-score MIN_PATH_SCORE
Minimum total Balrog score for a path of ORFs to be returned. [Default = 100]
--min-orf-score MIN_ORF_SCORE
Minimum individual Balrog score for an ORF to be returned. [Default = 100]
--max-orf-orf-distance MAX_ORF_ORF_DISTANCE
Maximum distance for graph traversal during ORF connection (bp). [Default = 10000]
--query-id QUERY_ID Ratio of query-kmers to required to match in graph. [Default = 0.8]
Settings to avoid/include algorithms:
--no-filter Do not filter ORF calls using Balrog. Will return all ORF calls. [Default = False]
--no-write-idx Do not write FMIndexes to file. [Default = False]
--no-write-graph Do not write Bifrost GFA and colours to file. [Default = False]
--repeat Enable traversal of nodes multiple times. [Default = False]
--no-clustering Do not cluster ORFs. [Default = False]
--no-refind Do not refind uncalled genes [Default = False]
Gene clustering options:
--identity-cutoff IDENTITY_CUTOFF
Minimum identity at amino acid level between two ORFs for clustering. [Default = 0.98]
--len-diff-cutoff LEN_DIFF_CUTOFF
Minimum ratio of length between two ORFs for clustering. [Default = 0.98]
--family-threshold FAMILY_THRESHOLD
protein family sequence identity threshold [Default = 0.7]
--merge-paralogs don't split paralogs[Default = False]
Panaroo run mode options:
--clean-mode {strict,moderate,sensitive}
R|The stringency mode at which to run panaroo. Must be one of 'strict', 'moderate' or 'sensitive'. Each of these modes can be fine tuned using the additional parameters in the 'Graph correction' section. strict: Requires fairly strong evidence (present
in at least 5% of genomes) to keep likely contaminant genes. moderate: Requires moderate evidence (present in at least 1% of genomes) to keep likely contaminant genes. sensitive: Does not delete any genes and only performes merge and refinding
operations. Useful if rare plasmids are of interest as these are often hard to disguish from contamination. Results will likely include higher number of spurious annotations.
Panaroo gene cluster annotation options:
--annotation {none,fast,sensitive,ultrasensitive}
Annotate genes using diamond default (fast), diamond sensitive (sensitive) or diamond and HMMscan (ultrasensitive). Specify 'none' if annotation not required.Default = 'fast'
--diamonddb ANNOTATION_DB
Diamond database. Defaults are 'Bacteria' or 'Viruses'. Can also specify path to fasta file for custom database generation
--hmmdb HMM_DB HMMER hmm profile file. Default is Uniprot HAMAP. Can alsospecify path to pre-built hmm profile file generated using hmmbuild
--evalue EVALUE Maximum e-value to return for DIAMOND and HMMER searches during annotation[Default = 0.001]
--truncation-threshold TRUNCATION_THRESHOLD
Sequences in a gene family cluster below this proportion of the length of thecentroid will be annotated as 'potential pseudogene'[Default = 0.8]
Panaroo gene refinding options:
the distance in nucleotides surronding the neighbour of an accessory gene in which to search for it
--refind-prop-match REFIND_PROP_MATCH
the proportion of an accessory gene that must be found in order to consider it a match[Default = 0.2]
Panaroo graph correction stringency options:
--min-trailing-support MIN_TRAILING_SUPPORT
minimum cluster size to keep a gene called at the end of a contig
--trailing-recursive TRAILING_RECURSIVE
number of times to perform recursive trimming of low support nodes near the end of contigs
--edge-support-threshold EDGE_SUPPORT_THRESHOLD
minimum support required to keep an edge that has been flagged as a possible mis-assembly
--length-outlier-support-proportion LENGTH_OUTLIER_SUPPORT_PROPORTION
proportion of genomes supporting a gene with a length more than 1.5x outside the interquatile range for genes in the same cluster.Genes failing this test will be re-annotated at the shorter length[Default = 0.1]
--min-edge-support-sv MIN_EDGE_SUPPORT_SV
minimum edge support required to call structural variants in the presence/absence sv file
--no-clean-edges Turn off edge filtering in the final output graph.[Default = False]
Alignment options:
--alignment {core,pan}
Output alignments of core genes or all genes. Options are 'core' and 'pan'. [Default = 'None'
--aligner {def,ref} Specify an aligner. Options: 'ref' for reference-guided MSA and 'def' for default standard MSA
--core-threshold CORE
Core-genome sample threshold.[Default = 0.95]
--no-variants Do not call variants using SNP-sites after alignment.[Default = False]
--ignore-pseduogenes Ignore ORFs annotated as 'potential pseudogenes' in alignment[Default = False]
Misc. options:
--balrog-db BALROG_DB
Path to save BALROG and default annotation databases. If not specified will downloadautomatically on first run.[Default = None]
--quiet suppress additional output[Default = False]
--threads THREADS Number of threads to use. [Default = 1]
--version, -v show program's version number and exit
実行方法
ゲノムのfastaファイルのリストを準備する。
cd fasta_dir/
ls -d -1 $PWD/*.fasta > input.txt
#dockerなら
ls -d -1 *.fasta > input_docker.txt
sed -i -e 's|^|/data/|' input_docker.txt
> cat input_docker.txt
![]()
準備ができたら実行する。dockerイメージを使う例
docker run --rm -it -v $(pwd):/workdir -v $(pwd):/data samhorsfield96/ggcaller:latest ggcaller --balrog-db /app/ggc_db --refs /workdir/input_docker.txt --out /workdir/ggc_out --threads 20
#dockerに入ってから使うなら
docker run --rm -it -v $(pwd):/workdir -v $(pwd):/data samhorsfield96/ggcaller:latest bash
cd /data/
ls -d -1 $PWD/*.fasta > input.txt
ggcaller -h
- --refs List of reference genomes (one file path per line).
- --reads List of read files (one file path per line).
- --threads Number of threads to use. [Default = 1]
10個のゲノムで15分ほどかかった(5995WX、10スレッド使用)。
出力例

cluster_size.png

summary_statistics.txt

gene_frequency.png
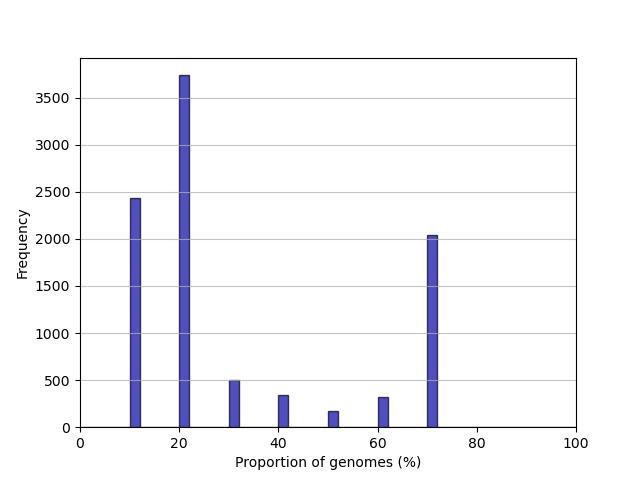
(あまり近くないゲノムをランダムに10個使用)
マニュアルより
- 多くのNを含むアセンブリは、アンダークラスタリングにつながるバラバラなDBGを生成する。最適なパフォーマンスを確保するため、Nsを含むアセンブリの使用は避ける。
- リードデータセット内(--reads)の遺伝子のコールはまだ広範囲にテストしていない。結果の解釈には注意する。
引用
Accurate and fast graph-based pangenome annotation and clustering with ggCaller
Samuel T Horsfield, Gerry Tonkin-Hill, Nicholas J Croucher, John A Lees
Genome Res. 2023 Sep;33(9):1622–1637. doi: 10.1101/gr.277733.123
関連