第3世代シークエンシング技術は、高品質でウルトラロングリードを生成できることから人気を博している。さまざまなサンプルや複数のシーケンスプラットフォームから得られたデータセットを比較および包括的な解析に活用することは、生物学的メカニズムの解明やベンチマークベースラインの確立に不可欠である。しかし、既存のロングリード用ツールは、主に個々のサンプルの品質管理(QC)とリード処理に重点を置いているため、複数のデータセットのプロファイリングと比較が複雑になっている。また、データの比較や結果の可視化のためのツールがないため、バイオインフォマティクスの経験が浅い研究者には困難が伴う。さらに、適切なシークエンシングプラットフォームを選択するために重要なベンチマークベースラインは、マルチサンプルおよびマルチプラットフォーム間での比較解析と可視化を容易にする包括的なロングリードQC手法の開発が必要である。Python3ベースのコマンドラインツールであるGiraffeは、複数のサンプルとプラットフォームにわたる比較解析と可視化のために設計された。このツールは、DNAシークエンシングリードとダイレクトRNAシークエンシングリードのリード品質、シークエンシングバイアス、ゲノム領域のメチル化比率を評価できる機能を提供することで際立っている。異なる生物学的処理法(全ゲノム増幅とショットガン)、シークエスプラットフォーム(Oxford Nanopore TechnologyとPacific Biosciences)、組織(血液を含む腎臓骨髄と含まない腎臓骨髄)、生物学的複製(腎臓骨髄)の比較など、さまざまなシナリオでの応用を通じてGiraffeの有用性を実証した。さらに、Oxford Nanoporeのduplexリードは、ホモポリマーの同定とGCの均一性においてPacBio HiFiリードを上回り、全体的なリードの質は同等であることがわかった。
インストール
ubuntu22でテストした。
mamba create -n giraffe -c bioconda -c conda-forge python==3.9 samtools==1.17 minimap2==2.17 bedtools==2.30.0 -y
conda activate giraffe
pip install Giraffe-View
> giraffe -h
usage: giraffe [-h] {estimate,observe,gcbias,modbin} ...
A tool to help you assess the quality of long-read sequencing data.
positional arguments:
{estimate,observe,gcbias,modbin}
estimate Estimated accuracy, length, and GC content.
observe Observed accuracy, mismatch proportion, and homopolymer identification.
gcbias Relationship between GC content and sequencing depth.
modbin Average modification proportion at regional level.
optional arguments:
-h, --help show this help message and exit
> giraffe estimate -h
usage: giraffe estimate [-h] --input <file list> [--cpu <number>] [--plot]
optional arguments:
-h, --help show this help message and exit
--input <file list> input the file list
--cpu <number> number of CPU (default:10)
--plot results visualization
> giraffe observe -h
usage: giraffe observe [-h] --input <file list> --ref <reference> [--cpu <number>] [--plot]
optional arguments:
-h, --help show this help message and exit
--input <file list> input the file list
--ref <reference> input reference
--cpu <number> number of CPU (default:10)
--plot results visualization
> giraffe observe -h
usage: giraffe observe [-h] --input <file list> --ref <reference> [--cpu <number>] [--plot]
optional arguments:
-h, --help show this help message and exit
--input <file list> input the file list
--ref <reference> input reference
--cpu <number> number of CPU (default:10)
--plot results visualization
> giraffe modbin -h
usage: giraffe modbin [-h] --input <list> --pos <reference> [--cpu <number>] [--plot]
optional arguments:
-h, --help show this help message and exit
--input <list> input list of modificated file
--pos <reference> input position file with CSV format
--cpu <number> number of CPU (default:10)
--plot results visualization
実行方法
4つのコマンドがある。
1,estimate - 推定リード精度(Qスコア)、長さ、GC含量の計算
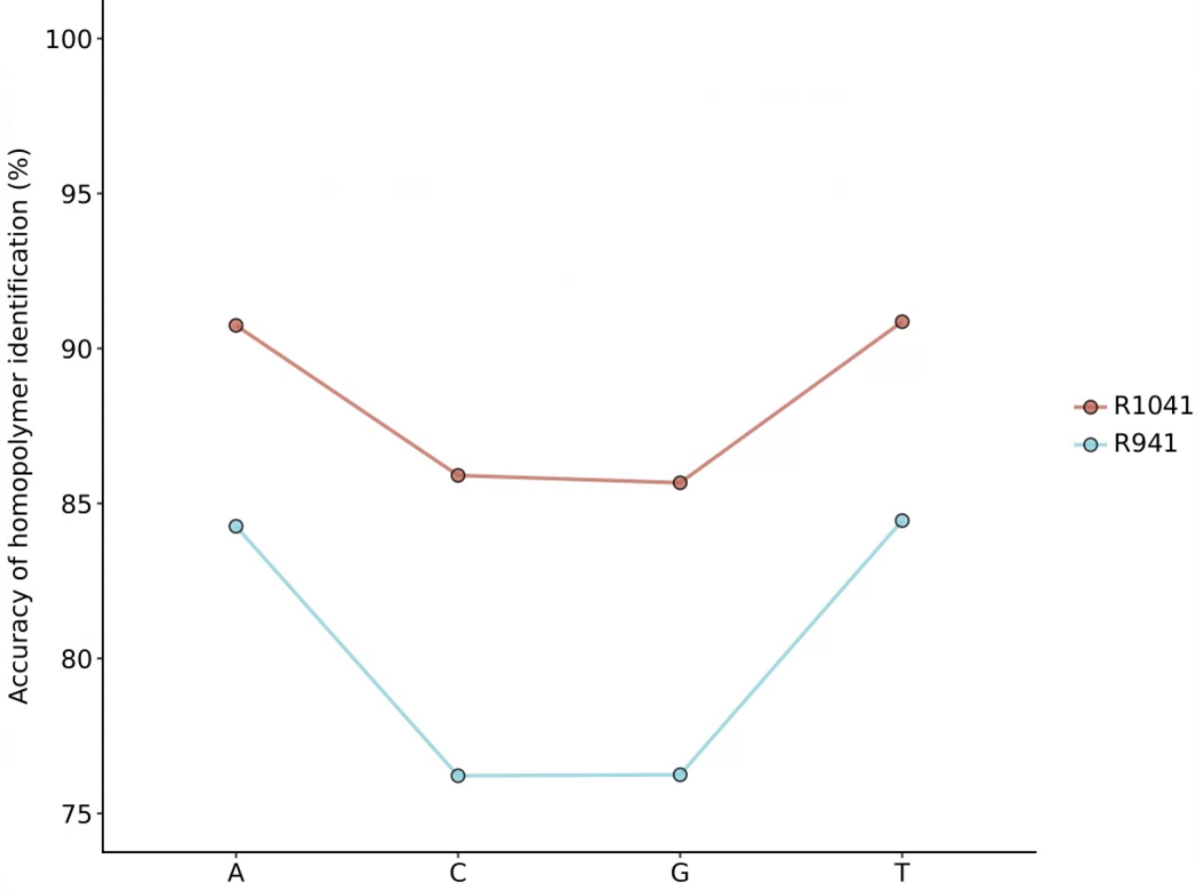
2,observe - 観測されたリード精度、ミスマッチの割合、ホモポリマーの同定(AAAAなど)の計算
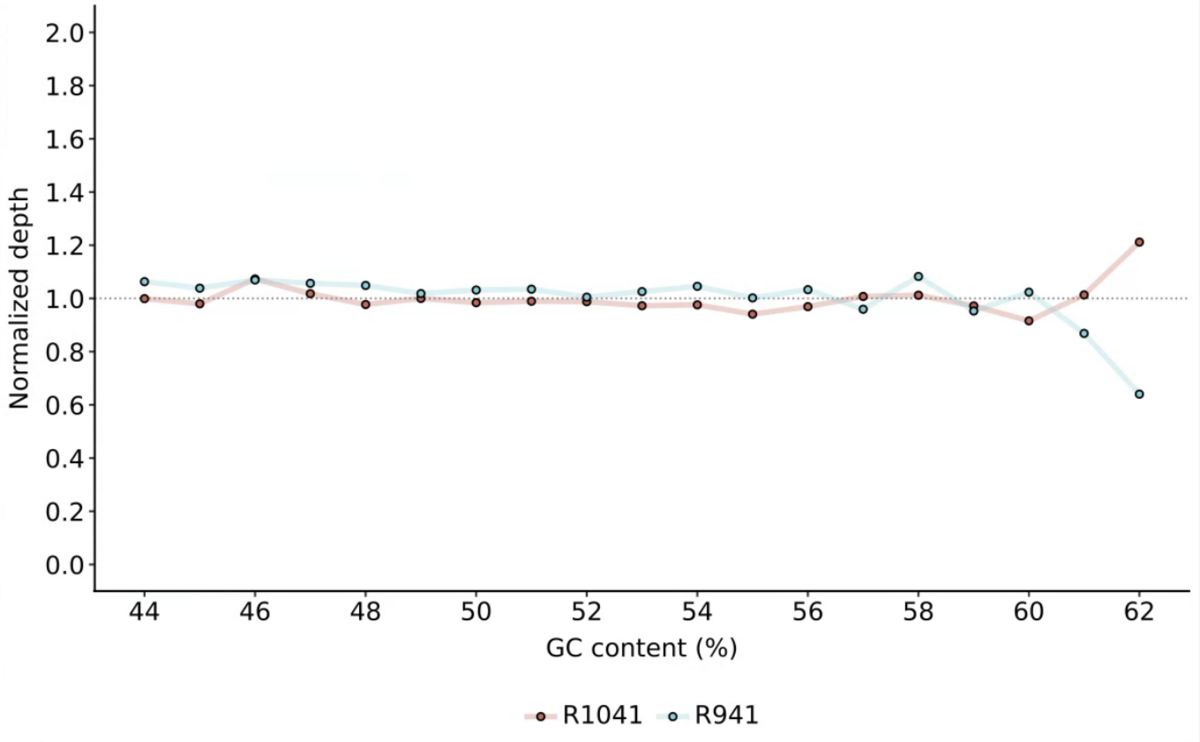
3,gcbias - GC含量とシーケンス深度の関係を計算
4,modbin - 領域レベルでの修飾(5mCや6mAメチル化など)の分布の計算
テストラン
run.shを実行する。
https://github.com/lrslab/Giraffe_View/blob/main/example/run.sh

run.shはexample下に含まれている。
git clone https://github.com/lrslab/Giraffe_View.git
cd Giraffe_View/example/
bash download.sh
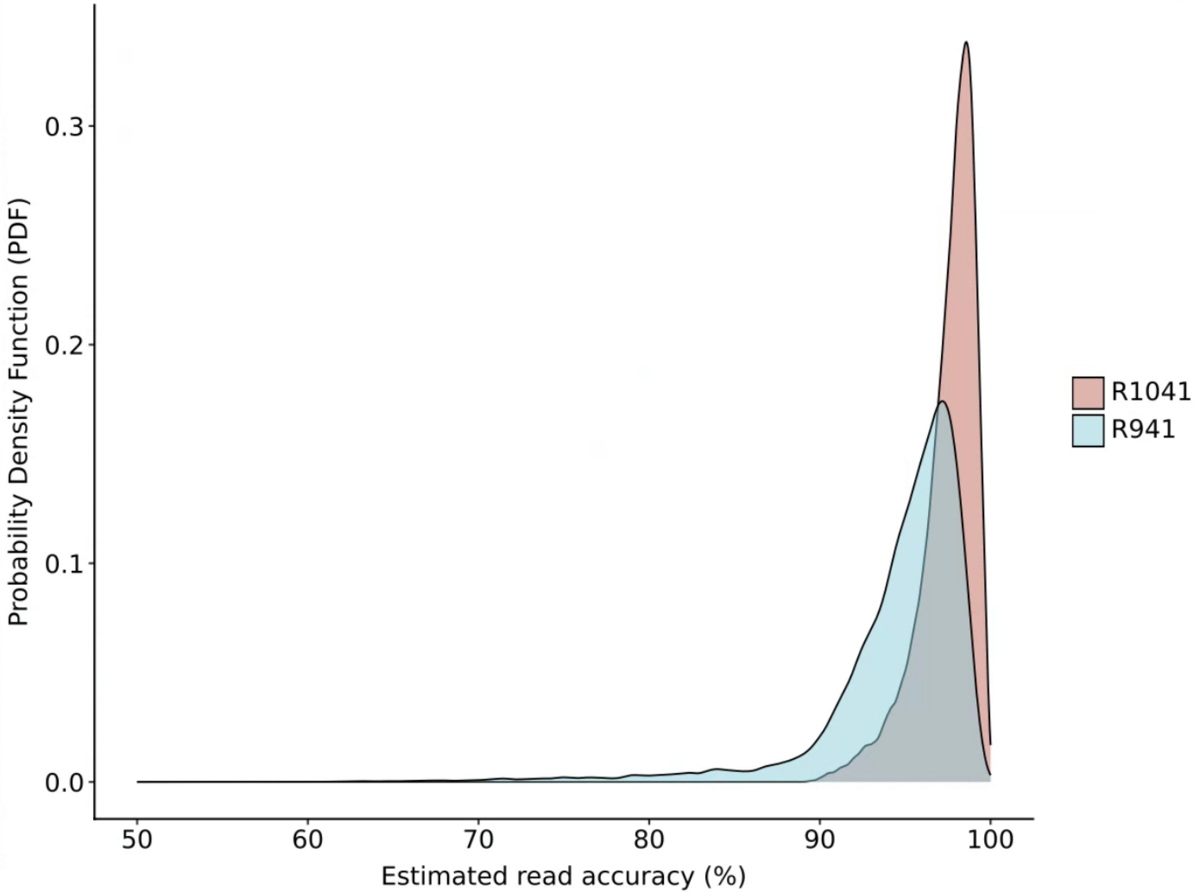
1、まずgiraffe estimateでリード精度(Qスコア)、長さ、GC含量の計算が実行される。
giraffe estimate --input fastq.list --plot --cpu 4
指定しているfastq.listにはfastqのパスが書かれている。
R941 ONT Read/R941.fastq
R1041 ONT Read/R1041.fastq
結果はGiraffe_Results/のサブディレクトリに保存される。
出力
1_Read_accuracy.pdf
2_Read_length.pdf

3_Read_GC_content

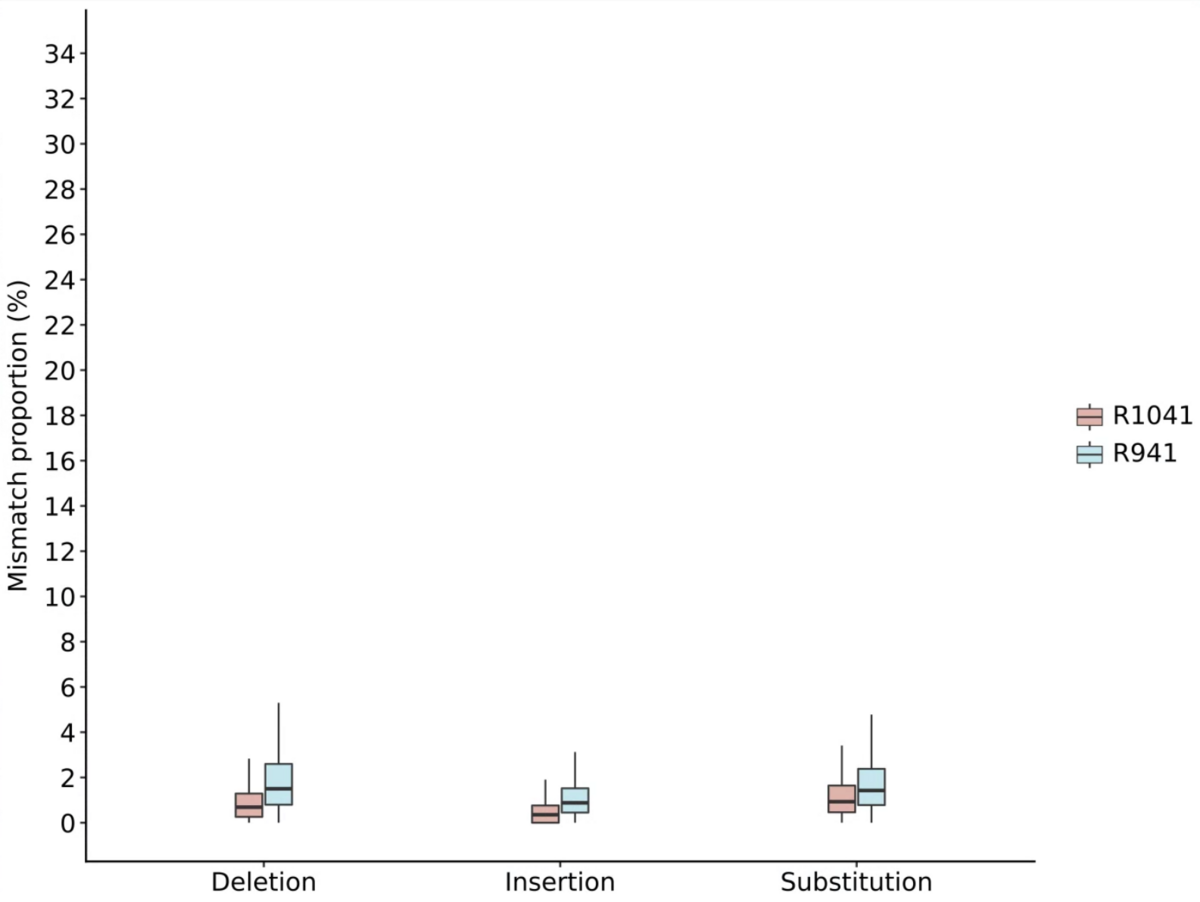
2、続いてobserveコマンドによって、リード精度、ミスマッチの割合、ホモポリマーの同定(AAAAなど)の計算が行われる。
giraffe observe --input fastq.list --plot --cpu 4 --ref Read/ecoli_chrom.fa
出力
2_Observed_mismatch_proportion.pdf
3_Homoploymer_summary.pdf
3、gcbiasでは、GC含量とシークエンスデプスとの関係が計算される。
giraffe gcbias --input bam.list --plot --ref Read/ecoli_chrom.fa
出力
1_Bin_distribution.pdf
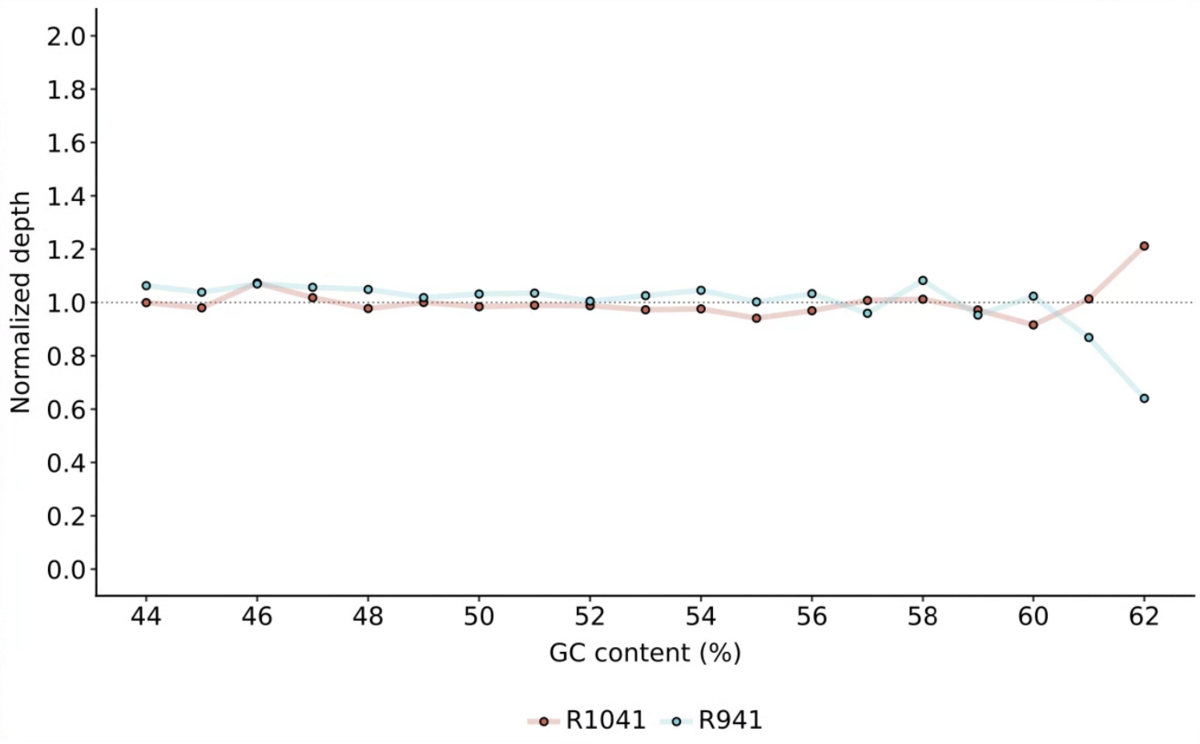
2_Relationship_normalization.pdf
4、最後に、modbinでは、塩基の修飾(5mCや6mAメチル化など)の分布が計算される。
giraffe gcbias --input bam.list --plot --ref Read/ecoli_chrom.fa
指定しているbam.listには、bamファイルのパスが1行ずつ書かれている。
cat bam.list
R941 ONT Giraffe_Results/2_Observed_quality/R941.bam
R1041 ONT Giraffe_Results/2_Observed_quality/R1041.bam
出力

コメント
- 出力内容についての詳細は、レポジトリで説明されています。確認して下さい。ここに結果を貼ったテストデータですが、さりげなくR10.4.1の方がR.9.4.1より品質が高いことを示す結果になってますね。
引用
Giraffe: a tool for comprehensive processing and visualization of multiple long-read sequencing data
Xudong Liu, Yanwen Shao, Zhihao Guo, Ying Ni, Xuan Sun, Yu Hung Anskar Leung, Runsheng Li
bioRxiv, Posted May 14, 2024.