2022/1/4 例追記,
2023/07/08 ツイート追記
2023/09/02 説明追加
以前紹介しましたが、分かりにくかったので、基本的な機能に限定して簡単にまとめ直します。
SnpEffはバリアントのアノテーションと機能的効果の予測ツールボックスです。遺伝的バリアントが遺伝子やタンパク質に与える影響(アミノ酸の変化など)をアノテーションし、予測します。SnpSiftは、データベースを利用してゲノム上のバリアントをアノテーションし、フィルタリングし、操作することができます。さらに、SnpEffを使ってファイルにアノテーションを付けたら、SnpSiftを使ってデータセットをフィルタリングし、その実験で最も重要なバリアントを絞り込むことができる(大規模なデータにも対応)。
HP
インストール
macos110.14でテストした。
依存
Building from the source
Most libraries should be install using Maven, so you just need to run mvn command.
#anaconda(link)
mamba create -n snpeff -y
conda activate snpeff
#snpeff
mamba install -c bioconda -y snpeff
#snpsift
mamba install -c bioconda -y snpsift
#dockerイメージ例(nf-core)
#v5.0.1
docker pull nfcore/snpeff:5.1.R64-1-1
#v.4.1イメージ例(biocontainers)
docker pull biocontainers/snpeff:v4.1k_cv3
> snpEff
$ snpEff
SnpEff version SnpEff 5.0e (build 2021-03-09 06:01), by Pablo Cingolani
Usage: snpEff [command] [options] [files]
Run 'java -jar snpEff.jar command' for help on each specific command
Available commands:
[eff|ann] : Annotate variants / calculate effects (you can use either 'ann' or 'eff', they mean the same). Default: ann (no command or 'ann').
build : Build a SnpEff database.
buildNextProt : Build a SnpEff for NextProt (using NextProt's XML files).
cds : Compare CDS sequences calculated form a SnpEff database to the one in a FASTA file. Used for checking databases correctness.
closest : Annotate the closest genomic region.
count : Count how many intervals (from a BAM, BED or VCF file) overlap with each genomic interval.
databases : Show currently available databases (from local config file).
download : Download a SnpEff database.
dump : Dump to STDOUT a SnpEff database (mostly used for debugging).
genes2bed : Create a bed file from a genes list.
len : Calculate total genomic length for each marker type.
pdb : Build interaction database (based on PDB data).
protein : Compare protein sequences calculated form a SnpEff database to the one in a FASTA file. Used for checking databases correctness.
seq : Show sequence (from command line) translation.
show : Show a text representation of genes or transcripts coordiantes, DNA sequence and protein sequence.
translocReport : Create a translocations report (from VCF file).
Generic options:
-c , -config : Specify config file
-configOption name=value : Override a config file option
-d , -debug : Debug mode (very verbose).
-dataDir <path> : Override data_dir parameter from config file.
-download : Download a SnpEff database, if not available locally. Default: true
-nodownload : Do not download a SnpEff database, if not available locally.
-h , -help : Show this help and exit
-noLog : Do not report usage statistics to server
-q , -quiet : Quiet mode (do not show any messages or errors)
-v , -verbose : Verbose mode
-version : Show version number and exit
Database options:
-canon : Only use canonical transcripts.
-canonList <file> : Only use canonical transcripts, replace some transcripts using the 'gene_id transcript_id' entries in <file>.
-interaction : Annotate using inteactions (requires interaciton database). Default: true
-interval <file> : Use a custom intervals in TXT/BED/BigBed/VCF/GFF file (you may use this option many times)
-maxTSL <TSL_number> : Only use transcripts having Transcript Support Level lower than <TSL_number>.
-motif : Annotate using motifs (requires Motif database). Default: true
-nextProt : Annotate using NextProt (requires NextProt database).
-noGenome : Do not load any genomic database (e.g. annotate using custom files).
-noExpandIUB : Disable IUB code expansion in input variants
-noInteraction : Disable inteaction annotations
-noMotif : Disable motif annotations.
-noNextProt : Disable NextProt annotations.
-onlyReg : Only use regulation tracks.
-onlyProtein : Only use protein coding transcripts. Default: false
-onlyTr <file.txt> : Only use the transcripts in this file. Format: One transcript ID per line.
-reg <name> : Regulation track to use (this option can be used add several times).
-ss , -spliceSiteSize <int> : Set size for splice sites (donor and acceptor) in bases. Default: 2
-spliceRegionExonSize <int> : Set size for splice site region within exons. Default: 3 bases
-spliceRegionIntronMin <int> : Set minimum number of bases for splice site region within intron. Default: 3 bases
-spliceRegionIntronMax <int> : Set maximum number of bases for splice site region within intron. Default: 8 bases
-strict : Only use 'validated' transcripts (i.e. sequence has been checked). Default: false
-ud , -upDownStreamLen <int> : Set upstream downstream interval length (in bases)
> snpEff databases -h
$ snpEff databases -h
Usage: snpEff databases [galaxy|html]
Options
galaxy : Show databases in a galaxy menu format.
html : Show databases in a HTML format.
> snpEff download -h
$ snpEff download -h
snpEff version SnpEff 5.0e (build 2021-03-09 06:01), by Pablo Cingolani
Usage: snpEff download [options] {snpeff | genome_version}
> snpsift
$ SnpSift
SnpSift version 4.3t (build 2017-11-24 10:18), by Pablo Cingolani
Usage: java -jar SnpSift.jar [command] params...
Command is one of:
alleleMat : Create an allele matrix output.
annotate : Annotate 'ID' from a database (e.g. dbSnp). Assumes entries are sorted.
caseControl : Compare how many variants are in 'case' and in 'control' groups; calculate p-values.
ccs : Case control summary. Case and control summaries by region, allele frequency and variant's functional effect.
concordance : Concordance metrics between two VCF files.
covMat : Create an covariance matrix output (allele matrix as input).
dbnsfp : Annotate with multiple entries from dbNSFP.
extractFields : Extract fields from VCF file into tab separated format.
filter : Filter using arbitrary expressions
geneSets : Annotate using MSigDb gene sets (MSigDb includes: GO, KEGG, Reactome, BioCarta, etc.)
gt : Add Genotype to INFO fields and remove genotype fields when possible.
gtfilter : Filter genotype using arbitrary expressions.
gwasCat : Annotate using GWAS catalog
hwe : Calculate Hardy-Weimberg parameters and perform a godness of fit test.
intersect : Intersect intervals (genomic regions).
intervals : Keep variants that intersect with intervals.
intIdx : Keep variants that intersect with intervals. Index-based method: Used for large VCF file and a few intervals to retrieve
join : Join files by genomic region.
op : Annotate using an operator.
phastCons : Annotate using conservation scores (phastCons).
private : Annotate if a variant is private to a family or group.
rmRefGen : Remove reference genotypes.
rmInfo : Remove INFO fields.
sort : Sort VCF file/s (if multiple input VCFs, merge and sort).
split : Split VCF by chromosome.
tstv : Calculate transiton to transversion ratio.
varType : Annotate variant type (SNP,MNP,INS,DEL or MIXED).
vcfCheck : Check that VCF file is well formed.
vcf2tped : Convert VCF to TPED.
Options common to all SnpSift commands:
-d : Debug.
-download : Download database, if not available locally. Default: true.
-noDownload : Do not download a database, if not available locally.
-noLog : Do not report usage statistics to server.
-h : Help.
-v : Verbose.
実行方法
1、データベースの有無の確認。
snpEff databases | grep -i Arabidopsis
出力
Arabidopsis_halleri Arabidopsis_halleri https://snpeff.blob.core.windows.net/databases/v5_0/snpEff_v5_0_Arabidopsis_halleri.zip
Arabidopsis_lyrata Arabidopsis_lyrata https://snpeff.blob.core.windows.net/databases/v5_0/snpEff_v5_0_Arabidopsis_lyrata.zip
Arabidopsis_thaliana Arabidopsis_thaliana https://snpeff.blob.core.windows.net/databases/v5_0/snpEff_v5_0_Arabidopsis_thaliana.zip
testAthalianaTair10 Test using athaliana's genome ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release https://snpeff.blob.core.windows.net/databases/v5_0/snpEff_v5_0_testAthalianaTair10.zip

2、データベースのダウンロード(リストにあってもダウンロードに失敗することがある)。
snpEff download -v Arabidopsis_thaliana
#バージョンで指定
snpEff download TAIR10.26
condaで導入した場合、データの保存場所は~/miniconda/envs/snpeff/share/snpeff-5.0-1/data/になる。各フォルダ中にはデータベースのバイナリファイル(.bin)が含まれる。
3、snpEffの実行。データベース名とvcfファイルを指定する。
snpEff -v -stats report.html Arabidopsis_thaliana variant_call.vcf > annotated.vcf
#バージョン名でダウンロードした場合
snpEff -v -stats report.html TAIR10.27 variant_call.vcf > annotated.vcf
公開されているシークエンシングデータを使用した。
出力
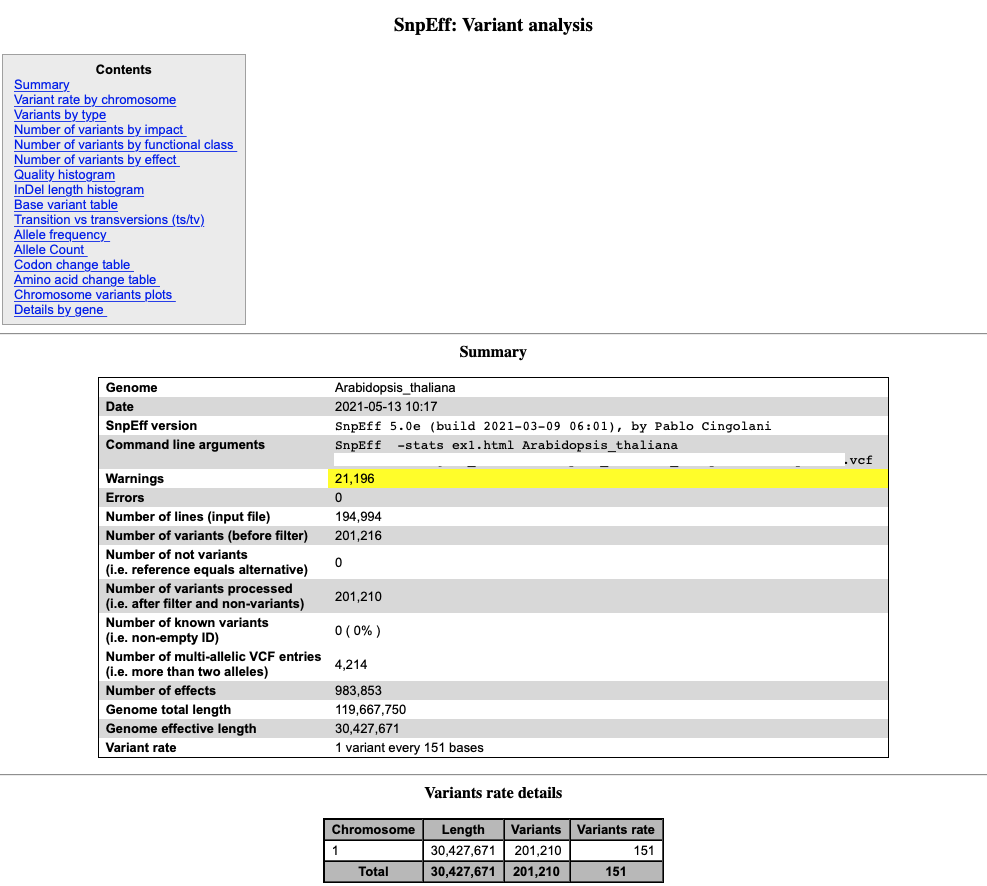
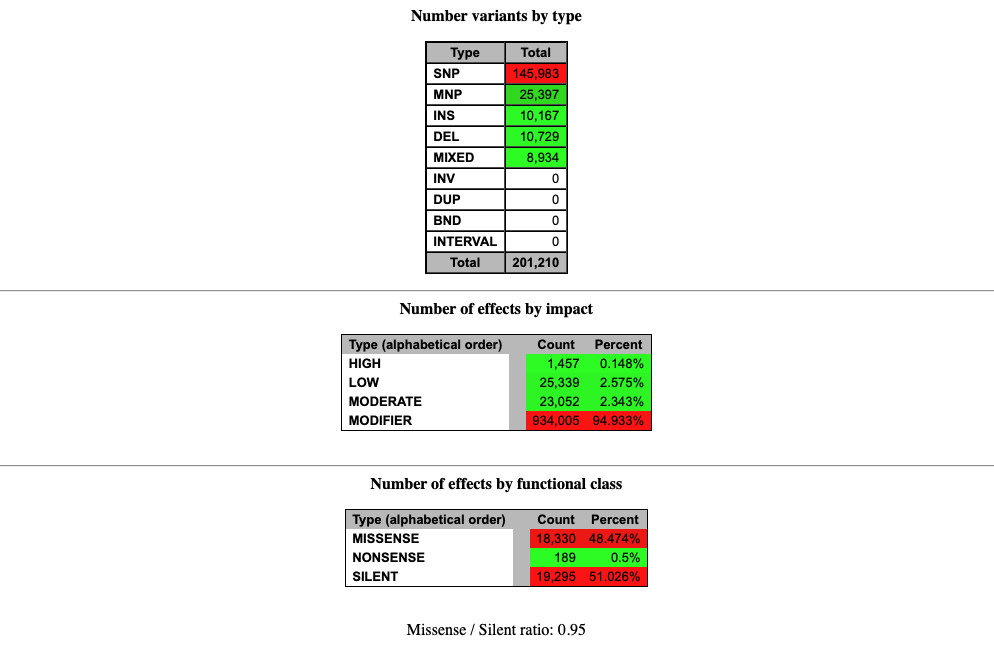
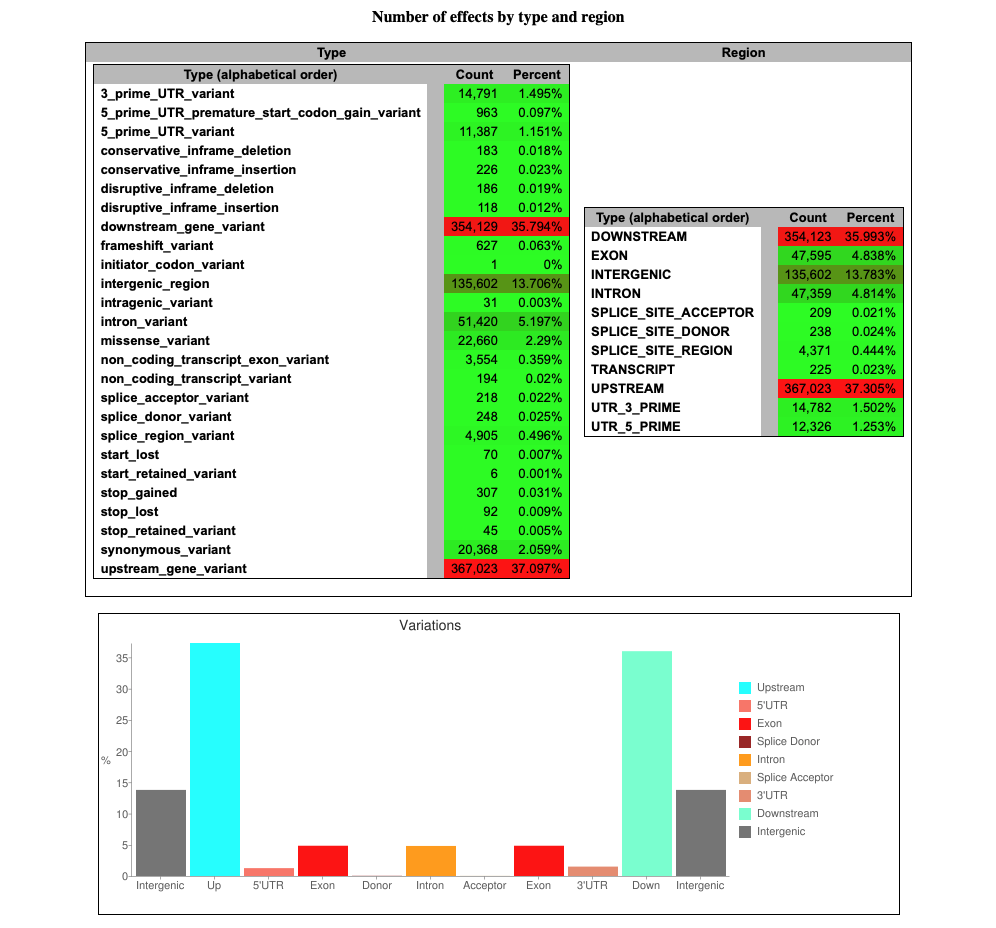
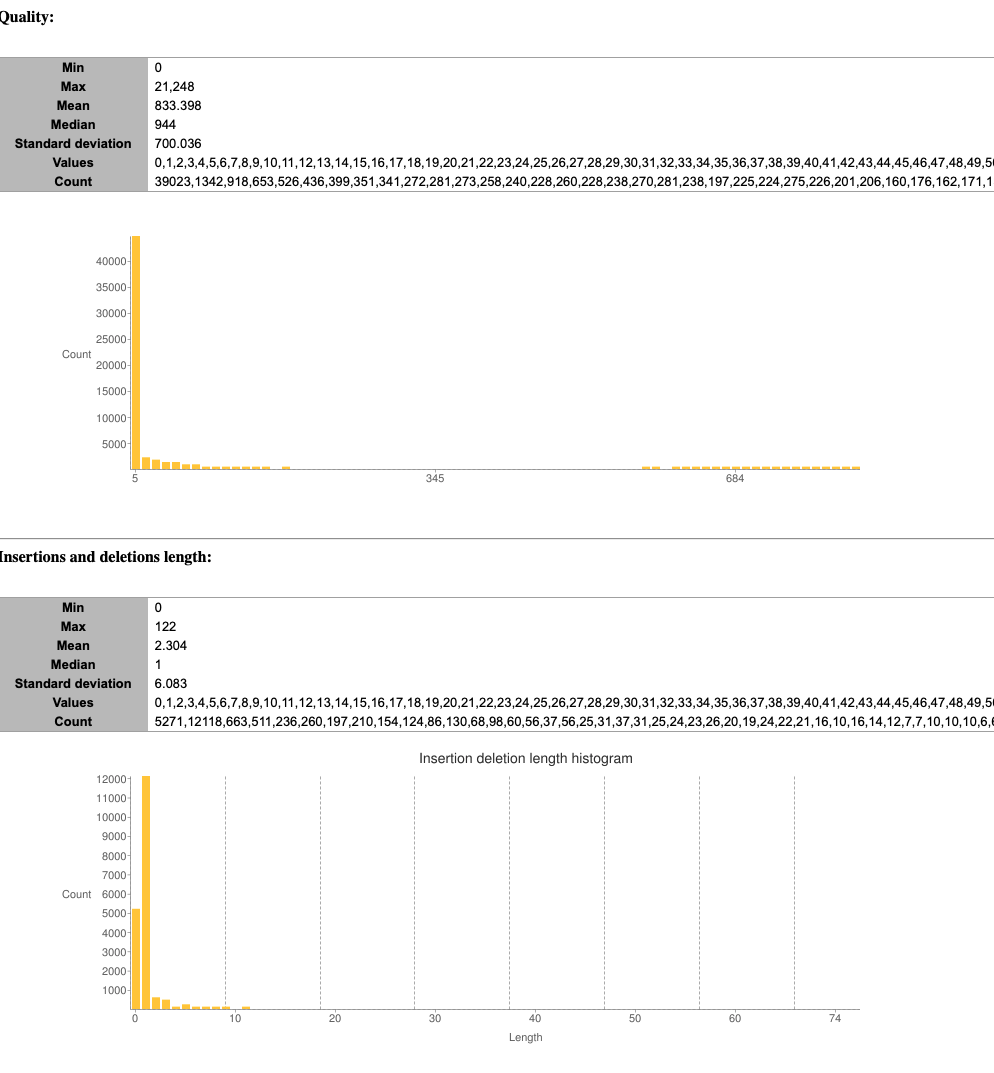

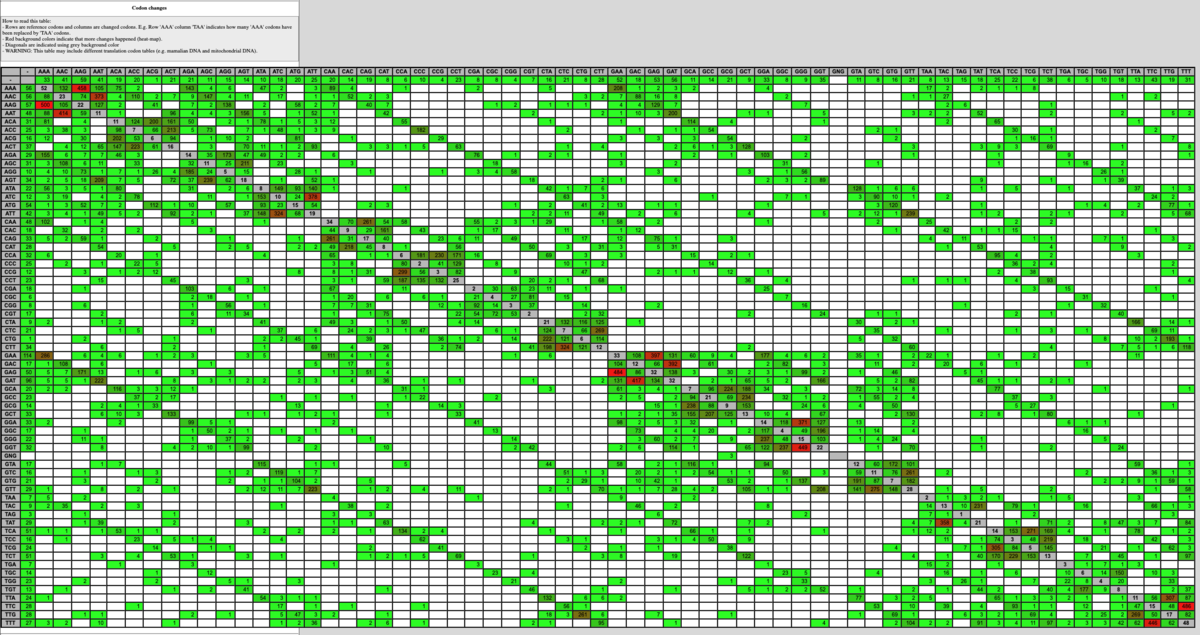
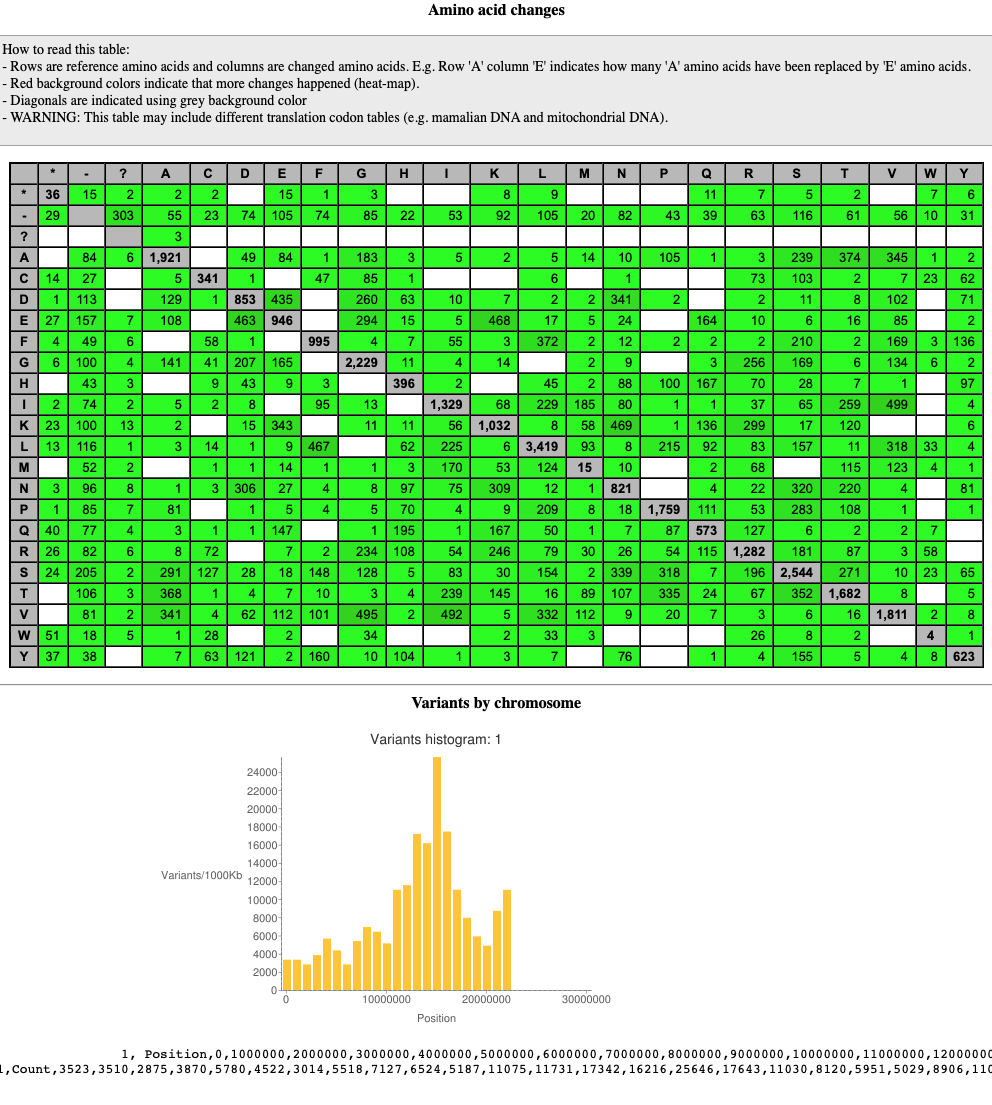
SnpEff は 3 つのファイルを出力する。
1、バリアントとそのアノテーションに関する要約統計を含むサマリーHTMLファイル(上の画像)。品質管理(QC)のためのmetricsとして利用できる。
2、アノテーションされたVCFファイル。
3、遺伝子ごとのバリアントタイプ数をまとめたテキストファイル。
Ts/Ts比やミスセンス/サイレント比がゲノム配列が決定された生物の期待値から大きく逸脱している場合は、ゲノム配列またはバリアントコールのパイプラインのいずれかに問題がある可能性がある。
カスタムアノテーション
データベースにないアノテーションデータも登録して使うこともできる。TXT, BED, BigBed, VCF, GFFなどを提供できるが、ここではexampleの解説に従って、GFFファイルからデータベースを作成する流れを確認する。
1、strainA.gffというGFFファイル(アノテーション+ゲノム配列)を使う。ディレクトリdata/strainAを作って、中に"genes.gff"という名前にリネームして収納する。
mkdir -p data/strainA
mv strainA.gff data/strainA/genes.gff
GFF3は末尾にゲノム配列を追加しておく必要がある。

(マニュアルより転載)
2、コンフィグファイルを作成する。
echo "strainA.genome : strainA" >> snpEff.config
カレントにsnpEff.configができる。

3、snpEff buildコマンドを使ってデータベースを作成する。カレントのコンフィグファイルが自動で認識される(-cで上書き)。gff3だけでなくgtfなども使える(アノテーションの後にリファレンスゲノム配列を結合したものが必要)。
snpEff build -c snpEff.config -gff3 strainA
Database format option (default: Auto detect):
- -embl : Use Embl format.
- -genbank : Use GenBank format.
- -gff2 : Use GFF2 format (obsolete).
- -gff3 : Use GFF3 format.
- -gtf22 : Use GTF 2.2 format.
- -knowngenes : Use KnownGenes table from UCSC.
- -refseq : Use RefSeq table from UCSC.
data/strrainA/ に.binファイルが保存される(デフォルトのローカルデータベースには保存されない)。
4、目的の機能(ここでは「ARS」機能)を持つファイルが必要。これらのfeatureは、オリジナルのGFFファイルに含まれているので、このファイルのfeature行だけを抽出する。
#まずGFFの配列が始まる行を調べ( grep -n "^#")、それより上の行xxx lineだけを保存する。
head -n xxx data/strainA/genes.gff > features.gff
5、snpEff の実行。features.gffファイル、configで設定した株(品種)の名前、ユーザーのVCFファイルを指定する。
snpEff -interval features.gff strainA my.vcf > annotated.vcf
2022/01/04
アノテーションをつける。メモリが足りなければ利用可能量を増やす。
java -Xmx64g -jar
~mambaforge/envs/snpeff/share/snpeff-5.0-1/snpEff.jar eff GRCh38.p13.RefSeq snps.vcf > snps_ann.vcf
variant effectについては公式ドキュメントを読んでください。一覧表が6ページ以降にあります。
variant annotations in VCF format
https://pcingola.github.io/SnpEff/adds/VCFannotationformat_v1.0.pdf
webチュートリアルについて
exampleでは、SnpEffとSnpSiftを使って、コーディングバリアントのアノテーション、優先順位付け、フィルタリングを行う方法が6つの例を使って説明されています。
1、Examples - SnpEff & SnpSift Documentation
SnpEffを使って入力VCFファイルの各バリアントレコードにアノテーションフィールドを追加し、フィルタリングプログラムであるSnpSiftを用いて、特定の基準を満たすアノテーションを持つ最も重要なバリアントを抽出します。CFTR遺伝子のナンセンス変異(G542*)というインパクトの強いコーディングバリアントを原因とするメンデル性劣性疾患、Cystic fibrosisの遺伝子変異を見つけるというものです。具体的には、Complete Genomics社が配列を決定した17人の健常者の血統から得られた7番染色体のバリアントコールのデータセットを使用しています。このデータセットでは、3人の兄弟に嚢胞性線維症の原因となるコーディングバリアントがこのデータ解析のために人為的に導入されています(リンク先の「資料」参照)。この例では、原因となるバリアントはわからないが、3人の兄弟は罹患しており(症例)、14人の両親と祖父母は罹患していない(対照)という、メンデル型劣性疾患を扱っていることはわかっています。そこで、症例のジョイントコールと対象のジョイントコールをそれぞれ行い、SnpEffでアノテーションします。血統がメンデルの劣性疾患と一致しているので、SnpSift caseControl コマンドで症例群と対照群における「ホモ接合」、「ヘテロ接合」、バリアントの数を数え(家系を指定するTFAMファイルを提供する)、Fisher exact 検定とCochran-Armitage 検定を用いて、いくつかの基本的なp値計算を行う。そして、SnpSift filterコマンドで 症例の3人全てホモ接合で、対照の誰もが変異のホモ接合ではないことを期待するフィルタリングを実行します。
2、シーケンス解析パイプラインで一般的に使用されるGATKとSnpEffを統合する方法と(SnpEffを使ってVCFファイルにアノテーションを付け、GATK VariantAnnotatorを使ってVCFファイルに最終的なアノテーションを組み込む)、Galaxy上でSnpEffを統合する方法が説明されています。
3、SnpEffとSnpSiftを使って、ノンコーディングバリアントのアノテーション、優先順位付け、フィルタリングを行う例です。先天性心疾患患者260人のT-box転写因子(TBX5)の遺伝子周辺700kbをシークエンスして同定された、20個のノンコーディングリストが使われています。推定転写因子結合部位(TFBS)を含めるようにしてSnpEffでのアノテーションを実行したが、ほとんどのバリアントが「INTERGENIC」としてカタログ化されており、候補となるSNVを絞り込むために使用できる追加情報は得られなかった(TFBSが小さいことや心臓や四肢の発生に関わる調節エレメントが、一般的に研究されている成人組織では広く活性化していない可能性)。そのため、他の研究のChIP-Seq解析で得られたデータ(BEDやBigBedなどで公開されていることも多い)を-intervalコマンドラインオプションで指定してsnpEffをランし、VCFにChIP-Seq実験の注釈を追加した(このコマンドラインオプションは、ENCODEプロジェクトやEpigenome RoadmapプロジェクトのChIP-Seq実験を使用してアノテーションを追加するのに利用できる。各コマンドラインで複数の-intervalオプションを使用できるので、複数のアノテーションを簡単に組み合わせることができる)。この-intervalオプションを指定した時に出力されるVCFには、bedファイルの指定されたゲノム領域と交差するバリアントに"CUSTOM[name of experiment]"という注釈が付く。それから、結果をSnpSift filterでフィルタリングして、制御領域で高度に保存されたSNPを探す、という流れです。
4、シーケンシングデータからアノテーションされたバリアントファイルを得る方法の例です(情報が古い)。
5、dbSnpにあるSNPをフィルタリングする例です。
引用
A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3
Pablo Cingolani 1 , Adrian Platts, Le Lily Wang, Melissa Coon, Tung Nguyen, Luan Wang, Susan J Land, Xiangyi Lu, Douglas M Ruden
Fly (Austin). Apr-Jun 2012;6(2):80-92
関連
snpeffのアノテーションの語彙
関連ツイート
2023/07/08
1/ Do you want to know if a variant will affect the structure and function of a protein?
— Ensembl (@ensembl) July 6, 2023
We’ve got lots of tools and #data in @ensembl to help you investigate and we’re here to show you how! A thread...🧵#genomics #bioinformatics # #tweetorial #Ensembltraining🧬 pic.twitter.com/PTM7fAcuAH