次世代シーケンシング(NGS)は、疾患に関連した遺伝的変異の発見を促進しており、疾患の病因調査および臨床遺伝診断に広く使用されている(Gong、Jiang、Duan、およびLu、2018; Zhao&Wei、2018)。 NGSによって識別された変異は、通常、変異データを格納するためのコミュニティ標準となっているVariant Call Format(VCF)ファイル(Danecek et al、2011)に格納される。 VCFは、1,000ゲノム(Genomes Project et al、2015)、ExAC(Lek et al。、2016)、ガンゲノムアトラス(TCGA)( Cancer Genome Atlas Research、2008)など、多くのゲノミクスプロジェクトで広く使用されている。VCFファイルの分析にはバイオインフォマティクスの専門知識が必要になる。 VCFのクエリとフィルタリングは、プログラミングスキルのない研究者にとって非常に困難である。バイオインフォマティクスの専門知識が不足しているセンターの医学遺伝学者は、バリアント解析のための使いやすいツールを見つけるのに苦労している。さらに、NGSから生成される突然変異データの量が指数関数的に増加しているため、これらのバリアントデータを管理するツールを開発することが急務になっている。
最近、バイオインフォマティクスのバックグラウンドが限られている研究者向けに、VCFファイルを処理するグラフィカルツールの開発に向けて多くの努力がなされている。 SNVerGUI(W. Wang、Hu、Hou、Hu、およびWei、2012)、database.bio(Ouなど、2015)、DaMold(Pandey、Pabinger、Kriegner、およびWeinhausel、2017)、mirVAFC(Li et al、2017)、GAVIN(van der Velde et al、2017)、およびgNOME(Lee et al、2014)は、非生物情報学者がバリアントの優先順位を決定するのを支援するために開発された。ただし、明確に定義されたアノテーションセットに依存し、VCFでユーザー定義のアノテーションを破棄し、ユーザーを事前定義された機能に制限するという欠点を共有している。経験豊富な研究者は、下流の分析のために社内のアノテーションを保持することを好むかもしれない。 VCF-Miner(Hart et al、2016)、VCF.Filter(Muller et al、2017)、myVCF(Pietrelli&Valenti、2017)、BrowseVCF(Salatino&Ramraj、2017)などのその他のツールは、アノテーションステップをスキップし、ユーザーがVCFで社内アノテーションを使用してバリアントをフィルタリングできるようにする。ただし、ほとんどの場合、ユーザーはバリアントを照会およびフィルタリングする前に、他のアノテーションツールでVCFにアノテーションを付ける必要がある。さらに、それらはインストールを必要とし、突然変異データを集中的に管理することはできない。
このギャップを埋めるために、バリアント分析用の高速で完全にインタラクティブなグラフィカルツールであるVCF-Serverを開発した。非バイオインフォマティクスの研究者は、プログラミングコードを1行も必要とせずに、VCFを照会し、バリアントにアノテーションを付けてフィルター処理することができる。 VCF-Serverを使用すると、ユーザーは一般的に使用されるデータベースまたはユーザー定義のバリアントアノテーション(バリアントブラックリストとホワイトリストを含む)でVCFにアノテーションを付けることができる。 VCFのバリアント情報は、インタラクティブなグラフィカルインターフェイスを介して視覚的に表示される。ユーザーは、柔軟なフィルタリングルールを使用してバリアントをフィルタリングできる。優先順位付けされたバリアントは、さらに分析するためにローカルコンピューターにエクスポートできる。 VCF-Serverは、VCFファイルを集中管理するためにWebファイルシステムを採用している。これにより、バリアント分析の効率が向上する。 VCF-Serverでは、直接的なWebベースの分析(デスクトップコンピューターまたはモバイルデバイスからアクセス可能)とローカル展開の両方が可能である。これにより、バイオインフォマティクスのバックグラウンドがほとんどない研究者が突然変異データを調査および解釈できるため、遺伝学分野でのトランスレーショナルリサーチが促進される。
VCF-Serverは、ブラウザー/サーバーアーキテクチャに基づくWebアプリケーションである。 VCF-Serverのコア機能モジュールはCで実装され、VCF処理を高速化する。 VCF-ServerのフロントエンドはSails.jsで記述され、Node.jsフレームワークで構築されている。バックエンドはPERL‐CGIで実装されている。 VCF-Serverは複数のユーザーをサポートし、すべてのユーザーが自分のプライベートデータ領域内で作業できるようにする。バックエンドからフロントに送信されるデータは、データ漏洩を防ぐために暗号化される。このプラットフォームは、単純なWebブラウザで完全に動作し、デスクトップコンピューターまたはモバイルデバイスのいずれかからアクセスできる。
manual
インストール
本体 Github
docker pull jiangjp880123/vcf-server
.vcf.gzファイルをdata/に保存して/vcf-serverのdockerイメージを立ち上げる。
cd path>/<to>/data
docker run -d -p 8000:9000 -v ${PWD}:/data jiangjp880123/vcf-server
http://127.0.0.1:8000にアクセスする。
実行方法
ここではオーサーらが用意しているデモサーバーを使いますが、ログインしないと全てのデータは他のユーザーからも見えているようです。mockデータ以外はdockerを使って下さい。
http://diseasegps.sjtu.edu.cn/VCF-Server?lan=eng にアクセスする。
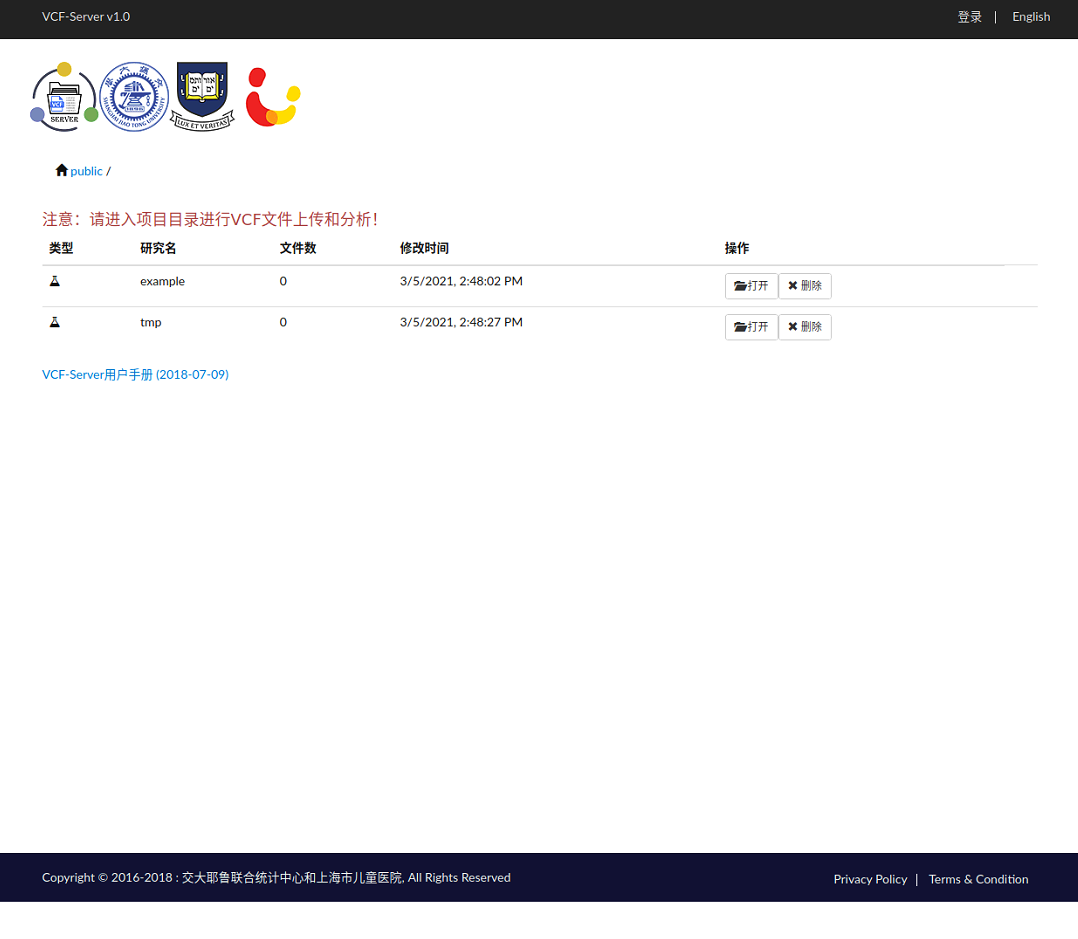
まず右上からEnglishに切り替えた。

exmapleのvcfファイルは7つ用意されている。
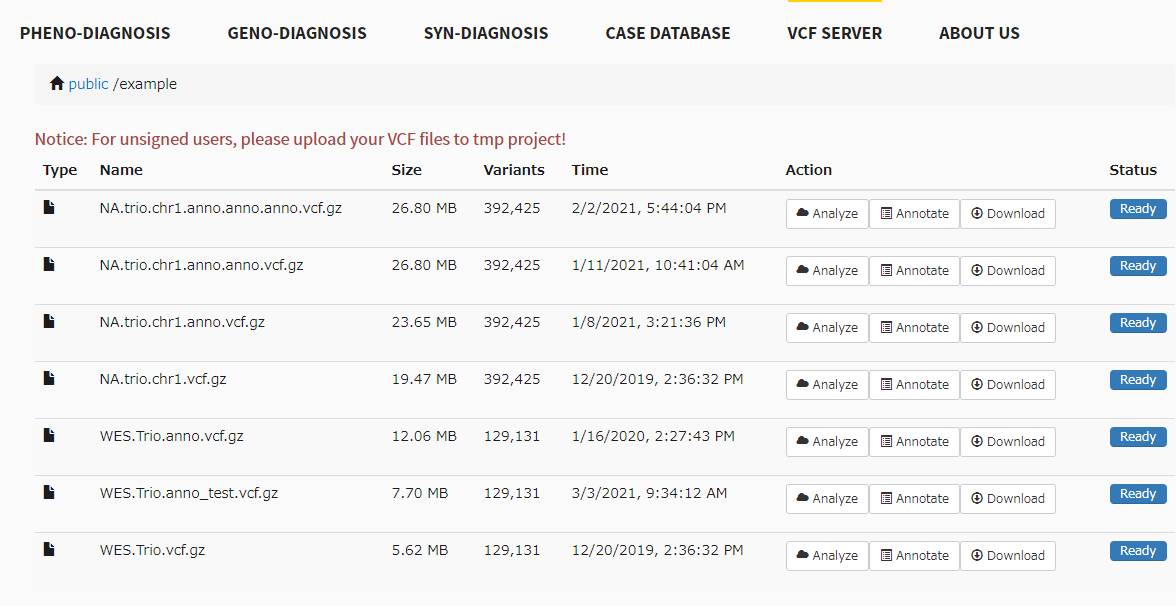
annotateボタンをクリック。ここではhg19のdbSNPでアノテーションを付ける。
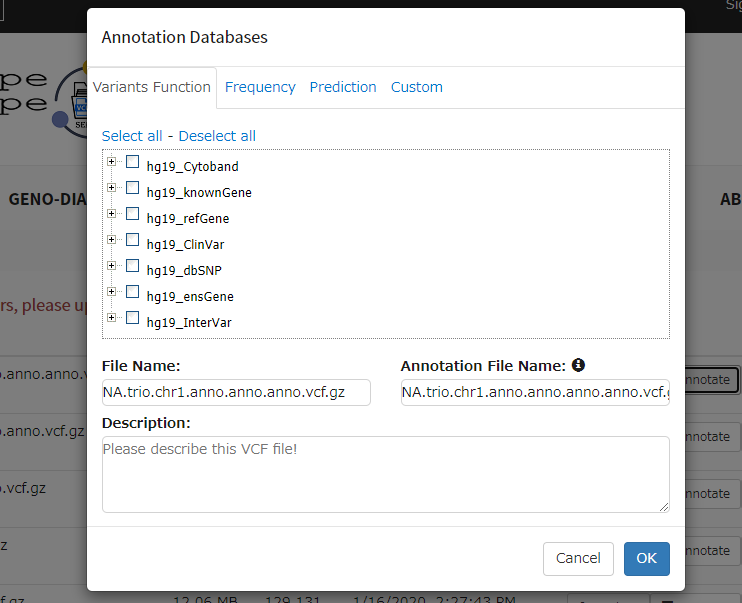
(すでにアノテーション済みのvcfを選んでしまっているため、出力はanno.anno.vcfという奇妙なファイル名になっている)
Annotation File Nameを決めて実行。しばらく時間がかかる(右端に進捗バーが表示される)。
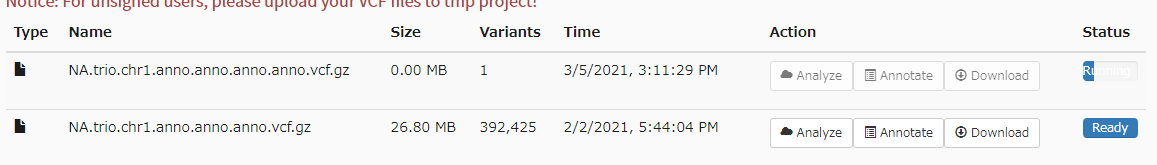
次にanalyzeをクリック。VCFを表で表示できる機能で、先にアノテーションをつけておけば、注釈カラムも表示することができる。

それにはDisplayボタンをクリックする。
表示項目を選ぶ。

IDの識別子がある場合、
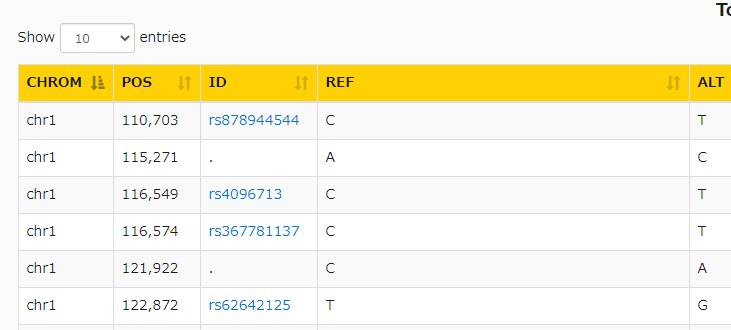
識別子はdbSNPにリンクしている。

Filterボタンをクリックすると、ユーザー指定の条件でバリアントコールのフィルタリングを実行できる(テスト時はフィルタリング条件が表示されなかった)。

上のタブからはバリアントについてサーチできる。

左からphenotypeをクリックして検索する。

引用
VCF-Server: A web-based visualization tool for high-throughput variant data mining and management
Jiang J, Gu J, Zhao T, Lu H
Mol Genet Genomic Med. 2019 Jul;7(7)