本著者らは、個体や集団のハイスループットな生シーケンスデータから遺伝子型バリアントコールまでのデータ処理を効率化するオールインワンSnakemakeワークフローであるgrenepipeを開発した。このパイプラインは、一般的なソフトウェアツールを単一の設定ファイル内で提供し、ソフトウェアの依存関係を自動的にインストールし、クラスタ環境でのスケーラビリティに高度に最適化され、単一のコマンドで実行できる。grenepipeはGPLv3の下で公開されており、github.com/moiexpositoalonsolab/grenepipeで自由に利用することができる。
https://github.com/moiexpositoalonsolab/grenepipe/wiki
Quick Start and Full Example
https://github.com/moiexpositoalonsolab/grenepipe/wiki/Quick-Start-and-Full-Example
インストール
依存
- conda(mamba含む)
- snakemake == 6.0.5
- python == 3.7.10
- pandas == 1.3.1
- numpy == 1.21.2
git clone https://github.com/moiexpositoalonsolab/grenepipe.git
cd grenepipe/
mamba env create -f envs/grenepipe.yaml -n grenepipe
conda activate grenepipe
実行方法
1、fastqの場所とサンプル名の関係を示したsample.tsvを用意する。フォーマットは、1列名はsample名、2列名は1回のシークエンシングデータなら1(複数ある場合は、下の画像のサンプルBのように2つ目のfastqには2をつける)、3列目はプラットフォーム名(GATKで影響)、4列名はfastqのパス(ペアエンドならforward fastq)、5列目はペアエンドのみでR2 (reverse) のfastqのパス。
https://github.com/moiexpositoalonsolab/grenepipe/wiki/Setup-and-Usage#samples-table
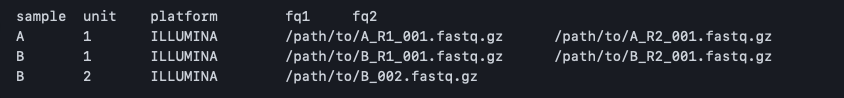
ここではsample.tsvとして保存する。
補足;tools/generate-table.pyを使うと、このサンプルTSVを自動で作成できる。fastqを含むディレクトリを指定する。
tools/generate-table.py <path>/<to>/fastq_dir
config.yamlの中でsample.tsvの場所を指定する。config.yamlのテンプレートはレポジトリの直下に用意されている。line13で作成したsample.tsvのパスを指定する。
https://github.com/moiexpositoalonsolab/grenepipe/blob/master/config.yaml
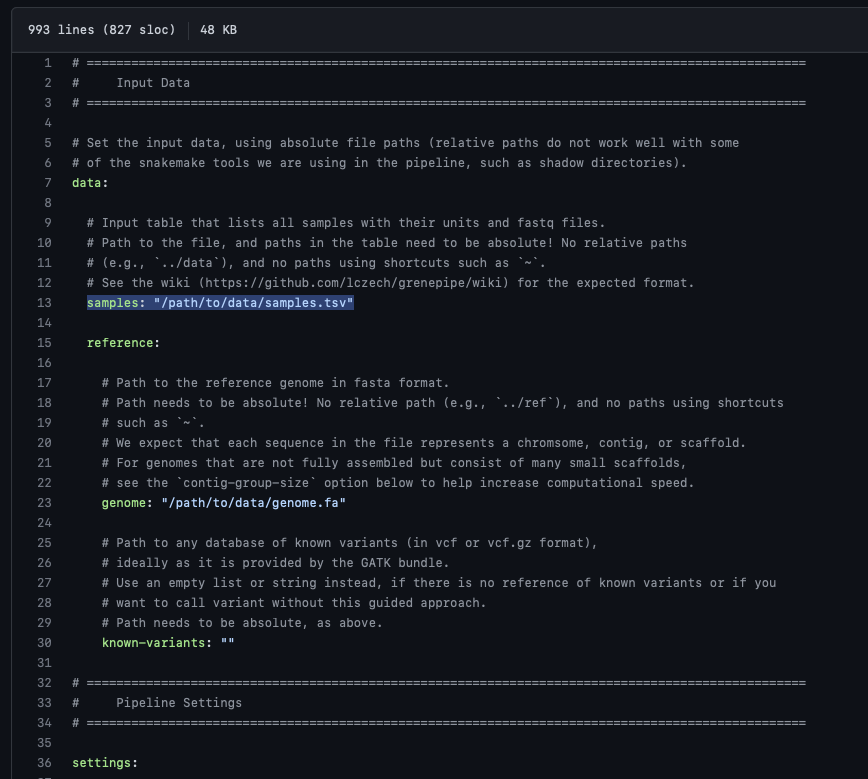
2、このconfig.yamlを結果を保存したい空のディレクトリに配置する。そのディレクトリのパスを指定して実行する。
snakemake --cores 20 --use-conda --directory /path/to/output
ディレクトリのYAMLファイルが認識され、パイプラインがスタートする。その後、envs/にある環境がインストールされるが、このインストールにかなり時間がかかる可能性がある。試したときはqualimap.yamlの環境導入でハングアップしていた(conda側の問題)。
引用
grenepipe: A flexible, scalable, and reproducible pipeline to automate variant calling from sequence reads
Lucas Czech, Moises Exposito-Alonso
Bioinformatics, Published: 02 September 2022