2021 10/24 conda -> mamba, 論文引用
2024/09/11 追記
NCBIは、タンパク質アノテーションおよび/またはアセンブルされたヌクレオチド配列を用いて、AMR遺伝子、抵抗性関連の変異、および他のクラスの遺伝子を同定するツールであるAMRFinderPlusを開発した。AMRFinderPlusは病原体検出パイプラインで使用され、これらのデータはNCBIのIsolate Browserに表示される。AMRFinderPlusは、NCBIのキュレーションされたリファレンス遺伝子データベースと隠れマルコフモデルのキュレーションされたコレクションに依存している。AMRFinderPlusの操作方法の詳細については、AMRFinderPlusドキュメントのMethodsセクションを参照してください。NCBIのすべての抗菌薬耐性リソースの説明については、ドキュメントを参照してください。
https://github.com/ncbi/amr/wiki
Isolates Browser (AMR phenotypeやserotype、isolation sourceなどを絞り込んでの病原菌の探索など。SRAにもリンクしている。)
https://www.ncbi.nlm.nih.gov/pathogens/isolates/#
インストール
mamba create -n ncbiamrfinderplus -y
conda activate ncbiamrfinderplus
mamba install -c bioconda ncbi-amrfinderplus -y
> amrfinder -h
$ amrfinder -h
Identify AMR and virulence genes in proteins and/or contigs and print a report
DOCUMENTATION
See https://github.com/ncbi/amr/wiki for full documentation
UPDATES
Subscribe to the amrfinder-announce mailing list for database and software update notifications:
https://www.ncbi.nlm.nih.gov/mailman/listinfo/amrfinder-announce
USAGE: amrfinder [--update] [--force_update] [--protein PROT_FASTA] [--nucleotide NUC_FASTA] [--gff GFF_FILE] [--pgap] [--database DATABASE_DIR] [--ident_min MIN_IDENT] [--coverage_min MIN_COV] [--organism ORGANISM] [--list_organisms] [--translation_table TRANSLATION_TABLE] [--plus] [--report_common] [--mutation_all MUT_ALL_FILE] [--blast_bin BLAST_DIR] [--name NAME] [--output OUTPUT_FILE] [--protein_output PROT_FASTA_OUT] [--nucleotide_output NUC_FASTA_OUT] [--quiet] [--gpipe_org] [--parm PARM] [--threads THREADS] [--debug] [--log LOG]
HELP: amrfinder --help or amrfinder -h
VERSION: amrfinder --version
OPTIONAL PARAMETERS:
-u, --update
Update the AMRFinder database
-U, --force_update
Force updating the AMRFinder database
-p PROT_FASTA, --protein PROT_FASTA
Input protein FASTA file
-n NUC_FASTA, --nucleotide NUC_FASTA
Input nucleotide FASTA file
-g GFF_FILE, --gff GFF_FILE
GFF file for protein locations. Protein id should be in the attribute 'Name=<id>' (9th field) of the rows with type 'CDS' or 'gene' (3rd field).
--pgap
Input files PROT_FASTA, NUC_FASTA and GFF_FILE are created by the NCBI PGAP
-d DATABASE_DIR, --database DATABASE_DIR
Alternative directory with AMRFinder database. Default: $AMRFINDER_DB
-i MIN_IDENT, --ident_min MIN_IDENT
Minimum proportion of identical amino acids in alignment for hit (0..1). -1 means use a curated threshold if it exists and 0.9 otherwise
Default: -1
-c MIN_COV, --coverage_min MIN_COV
Minimum coverage of the reference protein (0..1)
Default: 0.5
-O ORGANISM, --organism ORGANISM
Taxonomy group. To see all possible taxonomy groups use the --list_organisms flag
-l, --list_organisms
Print the list of all possible taxonomy groups for mutations identification and exit
-t TRANSLATION_TABLE, --translation_table TRANSLATION_TABLE
NCBI genetic code for translated BLAST
Default: 11
--plus
Add the plus genes to the report
--report_common
Report proteins common to a taxonomy group
--mutation_all MUT_ALL_FILE
File to report all mutations
--blast_bin BLAST_DIR
Directory for BLAST. Deafult: $BLAST_BIN
--name NAME
Text to be added as the first column "name" to all rows of the report, for example it can be an assembly name
-o OUTPUT_FILE, --output OUTPUT_FILE
Write output to OUTPUT_FILE instead of STDOUT
--protein_output PROT_FASTA_OUT
Output protein FASTA file of reported proteins
--nucleotide_output NUC_FASTA_OUT
Output nucleotide FASTA file of reported nucleotide sequences
-q, --quiet
Suppress messages to STDERR
--gpipe_org
NCBI internal GPipe organism names
--parm PARM
amr_report parameters for testing: -nosame -noblast -skip_hmm_check -bed
--threads THREADS
Max. number of threads
Default: 4
--debug
Integrity checks
--log LOG
Error log file, appended, opened on application start
> amr_report --help
$ amr_report --help
*** ERROR ***
"--help" is not a valid option
Report AMR proteins
USAGE: amr_report [-fam ""] [-blastp ""] [-blastx ""] [-gff ""] [-gff_match ""] [-bed] [-pgap] [-lcl] [-dna_len ""] [-hmmdom ""] [-hmmsearch ""] [-organism ""] [-mutation ""] [-mutation_all ""] [-suppress_prot ""] [-ident_min -1] [-coverage_min 0.5] [-skip_hmm_check] [-out ""] [-print_fam] [-pseudo] [-force_cds_report] [-non_reportable] [-core] [-name ""] [-nosame] [-noblast] [-nohmm] [-retain_blasts] [-qc] [-verbose 0] [-noprogress] [-profile] [-seed 1] [-threads 1] [-json ""] [-log ""] [-sigpipe]
HELP: amr_report -help
VERSION: amr_report -version
HOSTNAME: gw1
SHELL: /bin/bash
PWD: /home/kazumaxgene/normal/indepth_C05_MissingLibrary_1_HL5G3BBXX
Progam name: amr_report
Command line: amr_report --help
> amrfinder_update -h
$ amrfinder_update -h
Update the database for AMRFinder from https://ftp.ncbi.nlm.nih.gov/pathogen/Antimicrobial_resistance/AMRFinderPlus/database/
Requirements:
- the data/ directory contains subdirectories named by "minor" software versions (i.e., <major>.<minor>/);
- the "minor" directories contain subdirectories named by database versions.
USAGE: amrfinder_update [--database DATABASE_DIR] [--force_update] [--quiet] [--threads THREADS] [--debug] [--log LOG]
HELP: amrfinder_update --help or amrfinder_update -h
VERSION: amrfinder_update --version
OPTIONAL PARAMETERS:
-d DATABASE_DIR, --database DATABASE_DIR
Directory for all versions of AMRFinder databases
Default: /lustre7/home/kazumaxgene/miniconda3/envs/ncbiamrfinderplus/bin/data
--force_update
Force updating the AMRFinder database
-q, --quiet
Suppress messages to STDERR
--threads THREADS
Max. number of threads
Default: 1
--debug
Integrity checks
--log LOG
Error log file, appended, opened on application start
データベースの準備
mkdir amrfinder_db
cd amrfinder_db
amrfinder -u
- -u Update the AMRFinder database
- -U Force updating the AMRFinder database
テストラン
データのダウンロード
curl -O https://raw.githubusercontent.com/ncbi/amr/master/test_dna.fa \
-O https://raw.githubusercontent.com/ncbi/amr/master/test_prot.fa \
-O https://raw.githubusercontent.com/ncbi/amr/master/test_prot.gff \
-O https://raw.githubusercontent.com/ncbi/amr/master/test_both.expected \
-O https://raw.githubusercontent.com/ncbi/amr/master/test_dna.expected \
-O https://raw.githubusercontent.com/ncbi/amr/master/test_prot.expected
amrfinderのラン
amrfinder --plus -p test_prot.fa -g test_prot.gff -O Escherichia > test_prot.got
出力(1ファイル)

"-O <ORGANISM NAME>"をつけると、common geneのコールが抑制され、SNVコールも行われる。
カットオフ感度を60%に変更。
amrfinder --plus -p test_prot.fa -i 60 -g test_prot.gff -O Escherichia > test_prot.got
#gennome
amrfinder --plus -n genome.fa -i 60 -O Escherichia > test_prot.got
#最近のバージョンではコマンドが少し変わっている。
amrfinder --plus -p V.parahaemolyticus_ATCC_17802.faa -i 0.6 > test_prot.got
-
-i Minimum proportion of identical amino acids in alignment for hit (0..1). -1 means use a curated threshold if it exists and 0.9 otherwise Default: -1
ループでのランと複数の結果の結合(マニュアルより)
for assembly in *.faa
do
base=$(basename $assembly .faa)
amrfinder -p $assembly --threads 8 --plus --name=$base > $base.amrfinder
done
head -1 $(ls *.amrfinder | head -1) > combined.tsv
grep -h -v 'Protein identifier' *.amrfinder >> combined.tsv
出力例
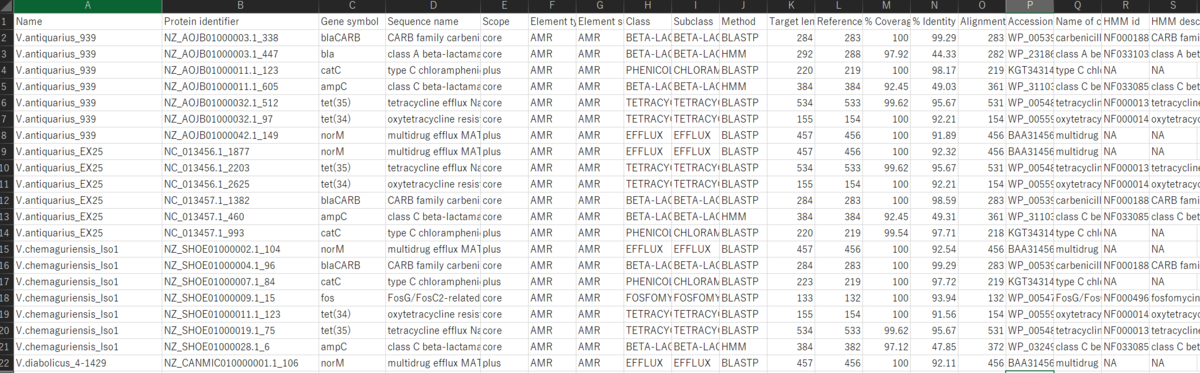
-Oで選択可能な生物は、2021年10月現在以下の通り
$ amrfinder --list_organisms
Available --organism options: Acinetobacter_baumannii, Campylobacter, Clostridioides_difficile, Enterococcus_faecalis, Enterococcus_faecium, Escherichia, Klebsiella, Neisseria, Pseudomonas_aeruginosa, Salmonella, Staphylococcus_aureus, Staphylococcus_pseudintermedius, Streptococcus_agalactiae, Streptococcus_pneumoniae, Streptococcus_pyogenes, Vibrio_cholerae
追記、2024年9月現在
Available --organism options: Acinetobacter_baumannii, Burkholderia_cepacia, Burkholderia_mallei, Burkholderia_pseudomallei, Campylobacter, Citrobacter_freundii, Clostridioides_difficile, Corynebacterium_diphtheriae, Enterobacter_asburiae, Enterobacter_cloacae, Enterococcus_faecalis, Enterococcus_faecium, Escherichia, Klebsiella_oxytoca, Klebsiella_pneumoniae, Neisseria_gonorrhoeae, Neisseria_meningitidis, Pseudomonas_aeruginosa, Salmonella, Serratia_marcescens, Staphylococcus_aureus, Staphylococcus_pseudintermedius, Streptococcus_agalactiae, Streptococcus_pneumoniae, Streptococcus_pyogenes, Vibrio_cholerae, Vibrio_parahaemolyticus, Vibrio_vulnificus
その他(マニュアルより)
- AMRFinderPlusを多数実行しようとすると、CPUが主なボトルネックになる。多数のジョブを並行して実行する場合に効率を最大化するには、各ジョブで--threads 1を使用することを推奨する(すべてのジョブを並行実行するのに十分なRAMがある前提)。
- AMRFinderPlus は /tmp にかなり大きなテンポラリファイルを読み書きするため、大規模に並列ランする場合は/tmp へのディスクアクセスがパフォーマ ンスに大きな影響を与える可能性がある。TMPDIR 環境変数を設定することで、一時ファイルの場所を変更することができる。
引用
https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/AMRFinder/
2021
AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence
Michael Feldgarden, Vyacheslav Brover, Narjol Gonzalez-Escalona, Jonathan G. Frye, Julie Haendiges, Daniel H. Haft, Maria Hoffmann, James B. Pettengill, Arjun B. Prasad, Glenn E. Tillman, Gregory H. Tyson & William Klimke
Scientific Reports volume 11, Article number: 12728 (2021)