多くの分類群において高品質な複数のリファレンスゲノム配列が利用可能になったことで、分子進化のパターンやプロセスを高解像度で見ることができるようになった。しかし、真核生物のほぼすべての系において、複数のリファレンスハプロタイプの情報を活用することは、依然として重要な課題である。このような課題は、染色体構造の進化の研究、量的形質座の候補遺伝子の探索、自然界における種分化や適応に関する仮説の検証など、多岐にわたる。ここでは、パンゲノムアノテーションの概念を通じてこれらの課題に取り組む。この概念では、保存された遺伝子順序を用いて遺伝子ファミリーを限定し、複数のゲノムアノテーションの中で共通の祖先を持つすべての遺伝子の予想される物理的位置を定義している。パンゲノムアノテーションを活用し、ゲノム間のシンテニックな関係を探ることにより、3種の4倍体綿花間、3億年にわたる脊椎動物の性染色体の進化、イネ科植物の多様性、トウモロコシ26品種間の存在・不在と構造変化を4段階の生物組織レベルで詳細に分析する。GENESPACE Rパッケージのシンテニックパン・ゲノムのアノテーションを構築し可視化する方法は、既存の遺伝子ファミリーおよびシンテニープログラム、特に倍数体、外来種、その他の複雑なゲノムを対象としたプログラムに大きな付加価値を提供するものである。
The preprint describing our comparative genomics #Rstats package is on bioRxiv (link below). GENESPACE uses synteny to explore variation among multiple genomes. For example, here are the syntenic regions to the human X and chicken Z sex chromosomes across 17 vertebrates pic.twitter.com/jrY0a4LWdp
— John Lovell (@_johntlovell) March 14, 2022
This approach works well with any ploidy. For example, here is a map of synteny across the grasses, including tetraploids (switchgrass and maize), and hexaploid wheat. Colors reflect synteny to the 12 rice chromosomes. pic.twitter.com/yhILpfGriY
— John Lovell (@_johntlovell) March 14, 2022
vignette
GitHub & BitBucket HTML Preview
インストール
mambaで環境を作ってorthofinderをインストール、MCScanXをビルド、GENESPACEとそれ以外のRのプログラムのインストールという順番で導入した。
mamba create -n orthofinder
conda activate orthofinder
mamba install -c conda-forge -c bioconda orthofinder -y
#MCScanX
git clone https://github.com/wyp1125/MCScanX.git
cd MCScanX/
make -j 8
export PATH=$PATH:$PWD
#RのコンソールでGENESPACEをインストール
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("jtlovell/GENESPACE", upgrade = F)
#bioconductorのrtracklayerとBiostringsも必要。インストールする
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("Biostrings", "rtracklayer"))
#もしくはcondaで導入できるものはcondaで導入する(残りはGENESPACEのみ)
mamba install -c conda-forge r-biocmanager -y
mamba install -c conda-forge r-devtools -y
mamba install -c conda-forge -c bioconda bioconductor-rtracklayer -y
mamba install -c conda-forge -c bioconda bioconductor-biostrings -y
#docker imageも出ている(おそらく非公式)
https://hub.docker.com/r/bcoli/genespace/tags
docker pull bcoli/genespace:1.0
テストラン
1、ライブラリをロードし、空のディレクトリ(ランデータ置き場)を指定する。ディレクトリは空ならO.K。存在していなくても問題ない。
#Rのコンソールにて
library(GENESPACE)
runwd <- file.path("./testGenespace")
2、ヒト/チンパンジー染色体3-4とアカゲザル染色体2および5のNCBIフォーマットアノテーションのサブセットがGENESPACE RパッケージのextDataで提供されている。
make_exampleDataDir(writeDir = runwd)
testGenespace/rawGenomes/が作成され、ヒト/チンパンジーのchr 3-4とアカゲザルのchr 2 & 5のNCBIフォーマットのアノテーションのサブセットが配置される。
testGenespace/rawGenomes/
 testGenespace/rawGenomes/chimp/chimp/annotation/
testGenespace/rawGenomes/chimp/chimp/annotation/

下流の柔軟性(例えば、1つの種に対して複数のゲノムバージョン、メタデータやアセンブリデータなど)のために、生ゲノムのディレクトリ構造は、/rawGenomes/[speciesID]/[versionID]/annotionとなっている。 アノテーションファイルは、"annotation "というサブディレクトリに格納される(マニュアルより)。
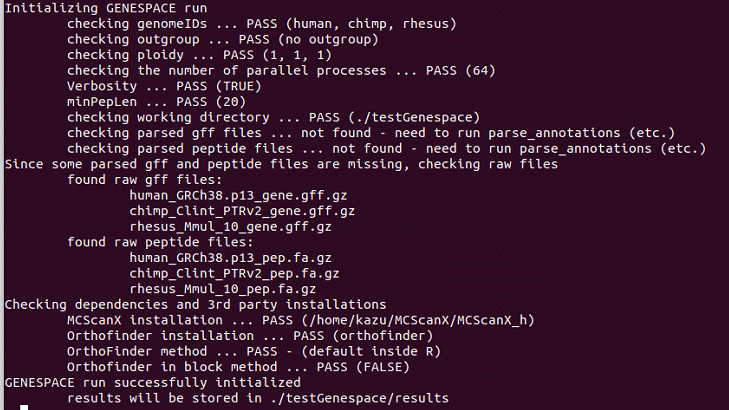
3、GENESPACEのランの初期化(vignetteの3参照)
プログラムのパス、などを指定する。
gids <- c("human","chimp","rhesus")
gpar <- init_genespace(
genomeIDs = gids,
speciesIDs = gids,
versionIDs = gids,
ploidy = rep(1,3),
wd = runwd,
gffString = "gff",
pepString = "pep",
path2orthofinder = "orthofinder",
path2mcscanx = "~/MCScanX",
rawGenomeDir = file.path(runwd, "rawGenomes"))
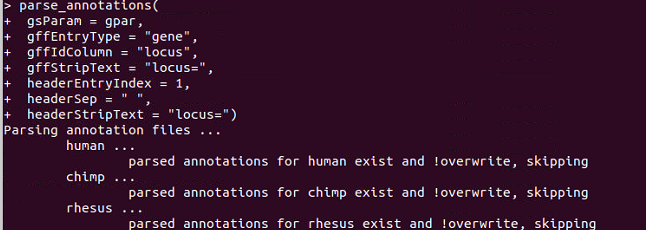
4、(マニュアルより)GENESPACEでは、ペプチドのFastaファイルのヘッダーとgff3アノテーションを正確にマージするために、ペプチドFastaヘッダーに正確に一致するカラムIDを持つ簡略化したgff3風テキストファイルが使用される。GENESPACEには、NCBI(parse_ncbi)およびphytozome(parse_phytozome)を解析する関数が組み込まれているが、非標準フォーマットのアノテーションを使用する場合、これは厄介なステップになる可能性がある。ここで使われているサンプルデータは元々NCBIからダウンロードしたものだが、ファイルサイズを小さくするため、NCBIのフォーマットを大幅に削除している。現在では、一般的なparse_annotationsでパースすることができる。
parse_annotations(
gsParam = gpar,
gffEntryType = "gene",
gffIdColumn = "locus",
gffStripText = "locus=",
headerEntryIndex = 1,
headerSep = " ",
headerStripText = "locus=")
5、orthofinderのラン
GENESPACEでは、orthofinderの実行が必要。orthofinderがパスに含まれているので、Rから直接、「高速」検索メソッドを使って実行できる(vignetteの5参照)。
gpar <- run_orthofinder(gsParam = gpar)

テストデータでは数十秒で計算は終わる。
6、synteny解析
GENESPACEのメインエンジンはsyntenyである。これは、ペアワイズブラストヒットをシンテニック領域とブロックに解析する複雑な関数となっている(vignetteの6,7参照)。解説ではデフォルト設定が使用されている。
gpar <- synteny(gsParam = gpar)

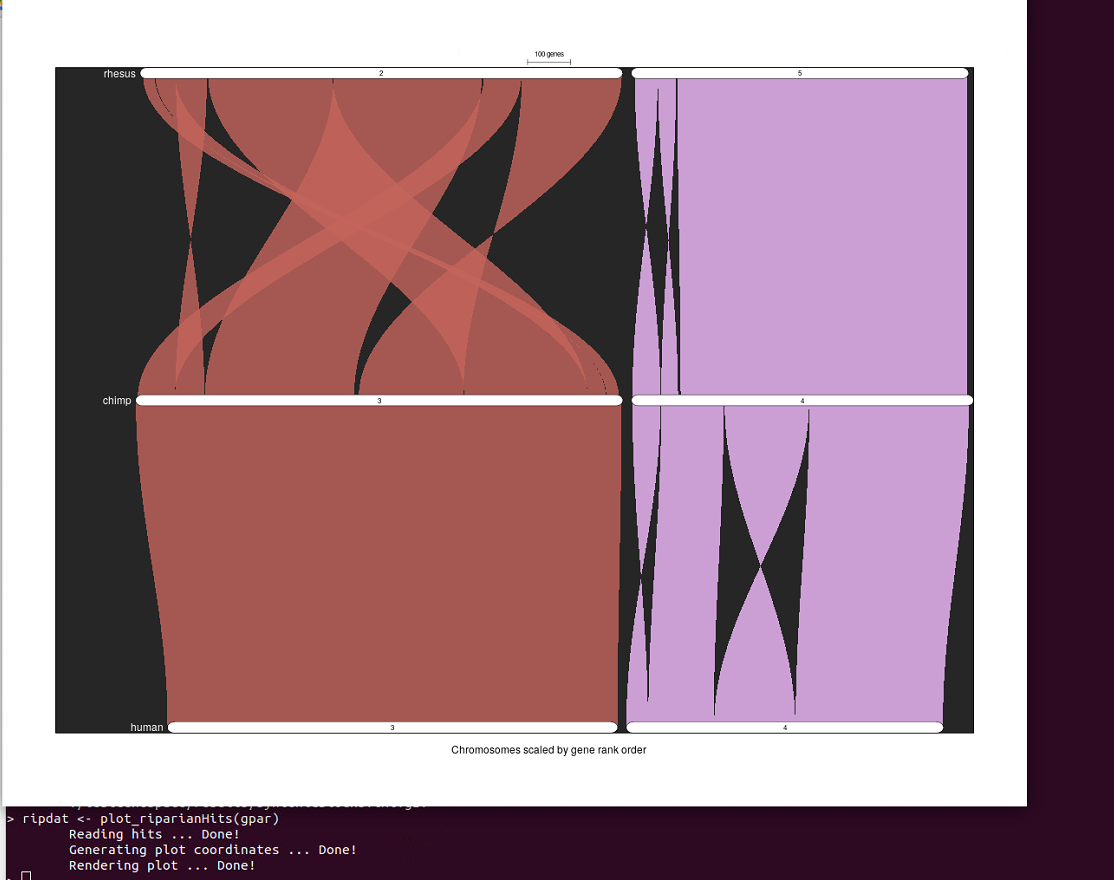
7、(マニュアルより)GENESPACEは、複数種のシンテニーを「リパリアン」プロットで可視化する。デフォルトでは、リファレンスゲノムに対するシンテニーの最大値で染色体を並べ、その起源となるリファレンス染色体によってシンテニーを色分けしている。このプロットは様々な方法でカスタマイズすることができる(vignetteの8.2参照、いくつかの例あり)。
ripdat <- plot_riparianHits(gpar)
8、パンゲノムアノテーションの構築
(マニュアルより)GENESPACEの主な出力はシンテニーアンカー付きのパンゲノムアノテーションで、シンテニーで制約された全てのorthogroupがリファレンスゲノム遺伝子オーダー上の位置に配置される。これはpangenome関数によって構築される(vignetteの8.2参照)。
pg <- pangenome(gpar)
(マニュアルより)GENESPACEは、リファレンスに対して構築された各パンゲノムについて、ファイルを書き出す。/results/[refgenome]_pangenome.txt.gz.というファイルを作成します。これは、プログラムで操作して目的の領域を抽出することができるソースデータに相当する。
各ステップで出力されるデータ

引用
GENESPACE: syntenic pan-genome annotations for eukaryotes
John T. Lovell, Avinash Sreedasyam, M. Eric Schranz, Melissa A. Wilson, Joseph W. Carlson, Alex Harkess, David Emms, David Goodstein, Jeremy Schmutz
bioRxiv, Posted March 11, 2022
関連
2022/06/14
Explore plant polyploidy with the new GENESPACE multi-scale synteny viewer on Phytozome. Go to https://t.co/xFIUPl1OIu or choose Synteny from the Phytozome Tools menu. @jgi @hudsonalpha @_johntlovell pic.twitter.com/YFuHrkI6ue
— Phytozome (@phytozome) June 11, 2022