2018 10/13 コマンドエラー修正
2019 11/13 インストール手順訂正
2021 5/9 docker image追記
全ゲノムシーケンシング戦略およびアセンブリアプローチの進歩により、一連の配列を互いに比較する方法が必要となっている。共通のクエスチョンは、同じリードセットの異なるアセンブリプログラムから得られたアセンブリ配列が互いにどのように異なっているか、または同じバクテリア種の異なる菌株のゲノムがどのように異なっているかである。このような分析を行うために、whole genome alignment(WGA)法がしばしば使用され、バイオインフォマティクスで長く研究されてきた。 WGAは、一般に、2つ以上の配列間の相同性の予測である[論文より ref.1]。 WGAは、主にゲノム間の保存された配列を同定するために使用される。ゲノム間の大規模な進化的変化を検出するゲノムの(functional)アノテーション、系統発生学的推論などをサポートする遺伝子、制御領域、非コードRNA配列、その他の機能要素[2,3]この分野は1970年代から継続的に発展しており、WGAのための多くの方法とツールが作成されている。既存の方法とツールのレビューは、[ref.1,4,5]にある。
シーケンスセット間の相違を検出する目的で、WGA解析を実行するために使用できるツールには特定の機能が必要になる。第一に、非常に断片化されたゲノム、構造上の再構成、ゲノム配列の複製、およびリピート領域に関連することが多い様々な相違に対処できるべきである。第二に、比較分析結果は、差異の種類およびその位置に関する情報を提供すべきである。この情報は、今後の分析に適した方法で保存される必要がある。そのような比較情報は、例えばリファレンスアシストのゲノムアセンブリ、アセンブリエラー検出、異なるアセンブリの比較のために使用することができる。第3に、さまざまなレベルで詳細なアラインメント結果を視覚化できるようにする必要がある。グローバル規模のビジュアライゼーションは、複製、再構成、カバーされていない領域を調べるために使用できるが、ローカルスケールのビジュアライゼーションでは、置換、挿入、欠失などの小さな違いに関する情報が得られる。
MAUVE [ref.6](紹介)、QUAST [ref.7]](紹介)、およびdnadiff [ref.8]の3つの異なるツールが現在利用できる。 MAUVEは、複数のゲノムアラインメントを行い、保存されたゲノム領域、これらの領域におけるリアレンジメントおよび逆位の正確なブレイクポイントを同定し、またヌクレオチド置換およびsmall indelsを同定する[ref.6]。また、インタラクティブな視覚化による結果の分析が可能で、情報を別々のファイルに保存する。しかし、小さな違い(置換、indels)に関する情報のみ、アクセサリプログラムを実行することなく簡単にアクセスできる。
QUASTは、ゲノムアセンブリの品質評価のためのツールであり、リファレンスゲノム存在下でアセンブリ品質に関する様々なmetricsを出力する。QUASTは構造変化のタイプとポジション情報のみ出力する。 QUASTは、Icarusと呼ばれる付随のゲノムブラウザでの可視化を可能にしている。しかし、QUASTは検出された違いのいかなる可視化も欠いており、要約統計のみを提供している。
Dnadiffは、MUMmer [ref.9]のNUCmerアラインメントプログラムのラッパーコマンドであり、相違を定量化し、アライメント統計と他の高レベルメトリクスを提供する[ref.8]。 QUASTと同様に、dnadiffはアセンブリの品質評価とゲノムの比較に使用できるが、検出された差を可視化することはできない。
ここでは、シーケンス比較のためにMUMmerのNUCmer、delta-filter、show-snpsプログラムを使用するNucDiffツールを紹介する。 NUCmerは配列同士をアライメントし、アライメントした領域に関する情報を出力する。これらの領域の相対的位置の厳密な分析により、ゲノムのリアレンジメントや逆位を含む様々なタイプの差異の検出を可能にし、場合によっては、それらの領域とリピート領域との関連性も確かめる。 NucDiffは、closely relatedな2つの配列の差異を特定し、その差異をいくつかのサブタイプに分類する。両方の入力シーケンスに関する全ての差異の正確な位置は、GFF3(Generic Feature Format version 3、[ref.10])ファイルとして出力される。正確なポジションにより、視覚化とさらなる分析の両方が可能になる。したがって、NucDiffによって提供される情報は、2つの配列セットがどのように異なるかを明らかにするのに役立つ。
NucDiffは、参照ゲノムおよびクエリーと呼ばれる2セットの配列間の様々なタイプの差異を、NUCmerによる配列のアライメントとdelta-filterおよびshow-snpsプログラムの結果を解析することによって決定する。 NUCmerはDNA配列アライメントを行い、デルタフィルタは指定された基準に従ってアライメント結果をフィルタリングする。デフォルトでNucDiffによって使用される設定では、delta-filterはクエリシーケンスの一貫性のある一番長いアラインメントを選択する。 NUCmer出力は、ソース配列、対応するリファレンスフラグメントに対するクエリフラグメントの方向、およびアライメントの類似性に関するすべてのフラグメントの正確な座標を含まれている。 show-snpsの結果には、対応リファレンスフラグメントと比較した、クエリフラグメント内のすべての挿入、欠失、および置換された塩基に関する情報が含まれる。

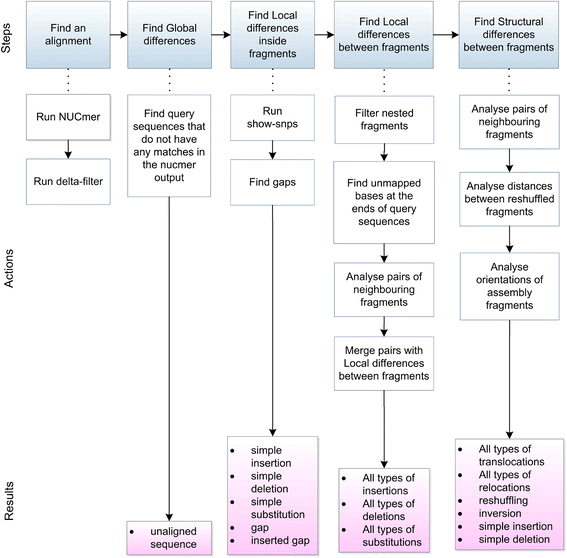
Classification of the types of differences. 論文より転載
著者らは、全ての変化は3つのグループに分けて考えることができると主張している。Nucdiffはそれぞれの違いを検出できるようパイプラインが構築されている(論文図2)。
{kind=link}
インストール
ubuntu18.04のpython3.5.1::Anaconda4.1.0でテストした。
依存
NucDiff can be run on Linux and Mac OS.
- Python 2.7
- MUMmer v3.23
- Biopython package
本体 GIthub
#ここでは仮想環境に入れる。python2.7の必要がある。
conda create -n nucdiff -c bioconda nucdiff -y
#bioconda (link)
mamba create -n nucdiff -c bioconda python==2.7.14 nucdiff -y
conda activate nucdiff
> nucdiff -h
$ nucdiff -h
usage: nucdiff [-h] [--reloc_dist [int]] [--nucmer_opt [NUCMER_OPT]]
[--filter_opt [FILTER_OPT]] [--delta_file [DELTA_FILE]]
[--proc [int]] [--ref_name_full [{yes,no}]]
[--query_name_full [{yes,no}]] [--vcf [{yes,no}]] [--version]
Reference.fasta Query.fasta Output_dir Prefix
positional arguments:
Reference.fasta - Fasta file with the reference sequences
Query.fasta - Fasta file with the query sequences
Output_dir - Path to the directory where all intermediate and
final results will be stored
Prefix - Name that will be added to all generated files
including the ones created by NUCmer
optional arguments:
-h, --help show this help message and exit
--reloc_dist [int] - Minimum distance between two relocated blocks
[10000]
--nucmer_opt [NUCMER_OPT]
- NUCmer run options. By default, NUCmer will be run
with its default parameters values, except the
--maxmatch parameter. --maxmatch is hard coded and
cannot be changed. To change any other parameter
values, type parameter names and new values inside
single or double quotation marks.
--filter_opt [FILTER_OPT]
- Delta-filter run options. By default, it will be run
with -q parameter only. -q is hard coded and cannot be
changed. To add any other parameter values, type
parameter names and their values inside single or
double quotation marks.
--delta_file [DELTA_FILE]
- Path to the already existing delta file (NUCmer
output file)
--proc [int] - Number of processes to be used [1]
--ref_name_full [{yes,no}]
- Print full reference names in output files ('yes'
value). In case of 'no', everything after the first
space will be ignored. ['no']
--query_name_full [{yes,no}]
- Print full query names in output files ('yes'
value). In case of 'no', everything after the first
space will be ignored.['no']
--vcf [{yes,no}] - Output small and medium local differences in the VCF
format
--version show program's version number and exit
——
ラン
クエリの配列と比較対象のリファレンス配列、出力、名前を指定して実行する。
nucdiff --vcf yes ref.fa query.fa outdir prefix
--vcf yesのフラグをつけると、SNV/indelについてvcfフォーマットでも出力される。
出力
resultsディレクトリに、リファレンスとクエリの各々から見たSNV、indel、構造変化それぞれの結果が出力される。

詳細はwikiに記載されています。
https://github.com/uio-cels/NucDiff/wiki
VCFやGFF3で出力されるので、そのままIGVに読み込んで可視化することもできる。

2021 5/10
以前動作しないというお話を伺ったので、イメージ(*1)をdockerhubにpushしておきます。
docker pull kazumax/nucdiff
cd <change>/<to>/<genome_dir>/
docker run --rm -itv $PWD:/data/ -w /data kazumax/nucdiff
nucdiff -h
> nucdiff
(nucdiff) root@6695c6ada349:/data# nucdiff
usage: nucdiff [-h] [--reloc_dist [int]] [--nucmer_opt [NUCMER_OPT]]
[--filter_opt [FILTER_OPT]] [--delta_file [DELTA_FILE]]
[--proc [int]] [--ref_name_full [{yes,no}]]
[--query_name_full [{yes,no}]] [--vcf [{yes,no}]] [--version]
Reference.fasta Query.fasta Output_dir Prefix
nucdiff: error: too few arguments
引用
NucDiff: in-depth characterization and annotation of differences between two sets of DNA sequences
Khelik K, Lagesen K, Sandve GK, Rognes T, Nederbragt AJ
BMC Bioinformatics. 2017 Jul 12;18(1):338.
関連ツール
*1
Dockerfile
FROM kazumax/miniconda:1.0 AS build-image
RUN mamba update -n base -c defaults conda
RUN conda create -n nucdiff -c bioconda python==2.7.14 nucdiff -y && mamba clean -a -y
RUN echo "conda activate nucdiff" >> ~/.bashrc
ENV PATH=/opt/miniconda3/envs/nucdiff/bin:$PATH
Dockerfileの場所で
docker build -t kazumax/nucdiff .
ベースイメージkazumax/miniconda:1.0はこちら