オペロンおよび機能的に連結された遺伝子アレイは原核生物ゲノムにおける転写構成の最も基本的な単位を表す。同じプロセスまたはパスウエイに関与する遺伝子は単一のブロックにコードされ、同じ調節の下で転写される。多くの臨床的に重要な遺伝子システムがオペロンにコードされている。すべての分泌システム、CRISPR-casシステム、Resistance Nodulation Division(RND)排出ポンプ、毒素抗毒素(TA)システムなどがこれに従っている。
同様の機能を有するオペロンおよび遺伝子アレイの構造は、単離体および種によって実質的に異なり得る。遺伝子の順序はしばしば変更され、個々の遺伝子は失われるかまたは獲得されるかもしれない。これらの違いはすべて、大規模データセットのゲノム間での比較を複雑にしている。これらの問題を解決するために、特定のオペロンにアノテーションを付ける洗練された方法が開発されてきた(ref.3,11–14)。これらのツールは、以前に定義された構造および配列に頼るか、または新しい遺伝的構造同定のために再プログラミングを必要とするので、特定のオペロンに限定される。あるいは、バクテリアゲノム中のすべてのオペロンを予測するためのツールが開発され、データベースを構築するために使用されてきた(ref.15–18)。これらのツールの多くはゲノムアノテーションファイルから検索するため、コーディングシーケンスが非常に短いために自動アノテーションプログラムで認識されないと、システムは観察されないままになる。重要な病原体のサーベイランスのため大規模データセットの利用可能性が高まっているため(ref.19–21)、幅広い分離株を臨床的に関連のある遺伝子アレイにアノテーションし、それらの多様性を調べるための単一の柔軟な枠組みが必要である。
ここではSLINGというLINKed Genesを検索するためのツールを紹介する。SLINGは、規則的に定義された近接性および方向性で、隣接配列とともに単一の保存された遺伝子として遺伝子配列を定義する。この定義により、SLINGは単離株にわたる遺伝子アレイの潜在的な多様性を捉えることができ、それらの多様性を同定し研究することができる。例えば、RND排出オペロンは常にRND排出ポンプタンパク質を含み、これはしばしば膜融合タンパク質の下流に位置する(ref.6)。毒素抗毒素(TA)システムでは、毒素タンパク質はその同族の抗毒素に近接してコードされている。SLINGを使用して、著者らは、(ref.22)から選択された70の腸病原性大腸菌(EPEC)ゲノムおよび選択された大腸菌参照株ゲノムからなる既存のデータセットにおいて、これら2つのオペロンを同定および特徴付けした。著者らは、単離系統学におけるこれらのシステムの分布ならびにそれらの遺伝的構成要素の多様性について洞察を得、特定の系統との関連を同定し、完全なアレイまたはそれらの構成要素全体にわたる構成要素の喪失または獲得のパターンについてより深い理解を得た。
https://github.com/ghoresh11/sling/wiki
インストール
mambaでpython2.7の環境を作ってテストした(計算にはubuntu18.04の計算機を使用)。
依存
sling has the following dependencies which need to be installed:
- python version >= 2.7.13
- HMMER version >= 3.1b2
- BLAST+ version >= 2.6.0
本体 Github
git clone https://github.com/ghoresh11/sling.git
cd sling
pip install .
> sling
$ sling
Usage: sling <command> <required arguments> [options]
To get minimal usage for a command use: sling command
To get full help for a command use one of: sling command -h OR sling command --help
Available commands:
run Run the complete search strategy on the input genomes
prepare Process the input genomes (in FASTA format, and optionally GFF format) for use by the following tasks.
scan Scan the ORFs from the preparation step using HMMER for profile collection
filter Summarise and filter the scanning results to identify hits that meet the structure requirements
group Group all hit and partner ORFs using sequence similarity networks
create_db Add custom HMM collection and requirements as built into SLING
view_dbs See list of avaiable built-in collections and their parameters
version Print version and exit
> sling run -h
$ sling run -h
usage: sling run [options] <run_id> <input_dir> <hmm_db>
Run the full search strategy
positional arguments:
PATH Directory to save all result files
PATH Name of directory containing FASTA files
STR/FILE Name of the predefined HMM database ['toxins',
'RND_pump'] OR path to custom HMM file
optional arguments:
-h, --help show this help message and exit
-fs STR, --fasta_suffix STR
Suffix of FASTA files in <input_dir> [.fasta]
-gd PATH, --gff_dir PATH
Name of directory containing GFF files. [<input_dir>]
-gs STR, --gff_suffix STR
Suffix of GFF files in <gff_dir> [.gff]
-mol INT, --min_orf_length INT
Minimun length of an open reading frame [20]
-c INT, --cpu INT Number of CPUs to be used [1]
-ct STR, --codon_table STR
Codon table to use in translation [Standard]
-sc STR, --start_codons STR
Accepted start codons written in hierarchical order of
usage [atg,gtg,ttg]
-o PATH, --out_dir PATH
Directory for all the output files
--hmmsearch STR HMM search executable (set to hmmscan if wish to run
scan not search) [Default: hmmsearch]
--hmmpress STR HMM press executable [relevant for hmmscan only]
[Default: hmmpress]
-u, --report_unfit Generate reports for HMMER hits that did not meet
requirements [False]
-mhl INT, --min_hit_length INT
Minimum length of a hit, if not in DOMAINS file [1]
-Mhl INT, --max_hit_length INT
Maximum length of a hit, if not in DOMAINS file
[10000000]
-mul INT, --min_upstream_length INT
Minimum length of the upstream gene [1]
-Mul INT, --max_upstream_length INT
Maximum length of the upstream gene [10000000]
-mdl INT, --min_downstream_length INT
Minimum length of the downstream gene [1]
-Mdl INT, --max_downstream_length INT
Maximum length of the downstream gene [10000000]
-Mo INT, --max_overlap INT
Maximum overlap between two operon proteins [300]
-Md INT, --max_distance INT
Maximum distance between two opern proteins [10000000]
-mhs FLOAT, --min_hmmscan_score FLOAT
Minimum HMMER score to use for significant hits [20]
-t STR, --order STR Location of partner gene relative to hit. Options:
upstream, downstream, either, both [either]
-s PATH, --sep PATH Delimiter to use in the output file [,]
-di FILE, --domains_to_ignore FILE
File with line delemited hmmer domains to ignore in
summary
-df FILE, --domains_file FILE
Tab delimited file of HMMER domains and the expected
length of their hits
-Mda INT, --max_diff_avg_length INT
Maximum difference between hit length and its average
length defined in <domains_file> [10000000]
-it, --save_to_ITOL Generate files that can be loaded into ITOL [False]
-mbe INT, --min_blast_evalue INT
Minimum BLAST evalue to use for an edge in the
sequence similarity network [0.01]
-mi INT, --min_identity INT
Minimum BLAST identity to use for an edge in the
sequence similarity network [30]
--makeblastdb STR makeblastdb executable [makeblastdb]
--blastp STR blastp executable [blastp]
実行方法
GFFファイル(full)を指定する。ITOLにロードするためのファイルも作成する。
sling run id genomes/ toxins -u --itol -c 12 -o outdir
- --itol flag states that files should be created to load into ITOL.
- -c Number of CPUs to be used [1]
-
-u Generate reports for HMMER hits that did not meet requirements [False]
-
-o Directory for all the output files
デフォルトではtozin antitoxinデータベースのHMMプロファイルが組み込まれており、このデータベースをtoxinsで選択している。u フラグをつけると廃棄されたヒットが報告される。
ステップ・バイ・ステップ
- prepare:検索用のFASTAファイル(およびオプションのGFFファイル)を処理する。
- scan: HMMERを使ってHMMプロファイルのためにORFをスキャンする。
- filter: スキャン結果をフィルタリングし、各ストレインにアノテーションを付ける。
- group:配列類似性ネットワークを使って、結果をグループ化する。
sling runは1-4を全て実行する。ステップ・バイ・ステップは、パラメータを変えながら、各ステップを繰り返しランする際に便利。
出力
出力内容についてはwikiで詳細に説明されています。wikiを確認して下さい。
--itolをつけてSLINGをランし、結果のid_GROUP/ITOL/のtxtファイル(wiki)をiTOLにドラッグして視覚化した。使用している系統樹ファイルは、(同じGFFから)roaryで連結コア遺伝子アラインメントを作成し、Fasttreeの近似法で系統推定することで得た。
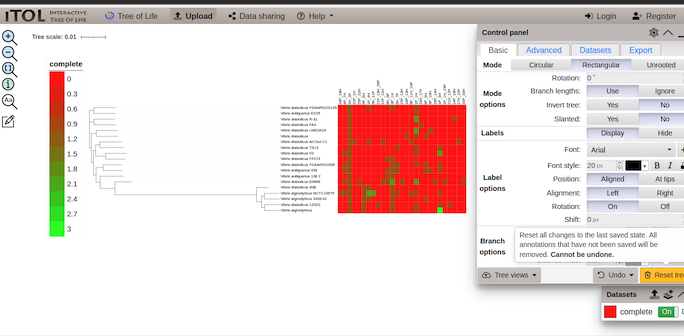
iTOLでは、系統樹ファイルを読み込ませたて可視化後、画面上にメタデータファイルをドラッグ&ドロップするとメタデータが視覚化される。ここではhits.txt(各ゲノムにおける各ヒットクラスタ数のmatrix)を読み込んでいる。同じデータはid_GROUP/のCSVファイルにもあり、ITOL/のtxtファイルはiTOL読み込み用となる。
引用
SLING: a tool to search for linked genes in bacterial datasets
Gal Horesh Alexander Harms Cinzia Fino Leopold Parts Kenn Gerdes Eva HeinzNicholas Robert Thomson
Nucleic Acids Research, Volume 46, Issue 21, 30 November 2018, Pages e128
関連