2023 5/30 タイトル修正
RNAシーケンスは、様々なRNAサブポピュレーションの発現に関する知識を得るために最も使用されるハイスループットなアプローチの1つとなっている。しかし、ライブラリー調製時やデータ解析時に発生する技術的なアーチファクトが、検出されたRNAの発現レベルに影響を与えることがある。特に、大規模かつ低インプットのデータセットや研究において重要なステップはデータの正規化であり、生物学とは関係のないデータのばらつきを排除することを目的としている。多くの正規化手法が開発されているが、それぞれが異なる仮定に依存しているため、適切な正規化戦略を選択することが生物学的情報を保存する上で重要となる。そこで著者らは、与えられたデータセットにおける正規化手法の性能を系統的に評価するための無料のウェブサーバーツール、NormSeqを開発した。NormSeqの主な特徴は、最適な正規化法の選択を導くinformation gainの実装であり、これは非生物学的変動を排除するか少なくとも減らすために重要である。NormSeqは、バイオインフォマティクスの専門知識がない研究者でも、データから信頼できる生物学的推論を得ることができるように、データの正規化に焦点を当てた遺伝子発現データの様々な側面を探求する使いやすいプラットフォームを提供する。NormSeqは、https://arn.ugr.es/normSeqで自由に利用できる。
help
https://arn.ugr.es/normseq_doc/
Very proud and happy to share our last tool NormSeq published at @NAR_Open.
— Cris Gómez (@cris12gm) 2023年5月22日
If you have RNA-seq data, do not hesitate to find the best normalization for your dataset at https://t.co/qLAdaYoQd7! It includes DE analysis, visualization options, and so on.https://t.co/9UsuXQyAV9
https://arn.ugr.es/normSeqにアクセスする。

Quick startを見てみる。
Quick startでは、マウスのtRNA発現データを使ってNormSeqプラットフォームについて学ことが出来る。

チュートリアルでは、GEOデータセットGSE141436のマウス組織サンプルからなるtRNA発現データセットが使われている。このデータセットは、中枢神経系(CNS)、肝臓、脛骨、心臓マウス組織から得られた7つのマウス組織からの21種類のサンプルの、tRNAの転写産物レベルでのtRNAシーケンス生カウント行列からなる。
データをロードする。

(発現)マトリックスファイルをロードする。発現マトリックスはアップロードするか、リンク/URLとして指定する。どちらの場合も、txt、csv、tsv、およびxlsの形式を認識する。

マトリックスファイルは、サンプル数分の列数と特徴数(遺伝子など)と同じ数の行数を持つrawリードカウントテーブルを指す。最初の列の1行目はnameとする。
チュートリアルのデータはURLからロードされる。

uploadからもデータは読み込める。

2つ目の入力ファイルとしてアノテーションマトリックスが必要。アノテーションマトリックスもURLとファイルどちらでも提供可能。どちらの場合も、txt、csv、tsv、xlsフォーマットが使用可能。

アノテーションマトリックスには2つの列が必要。1列目には入力マトリックスとサンプル名が必要で、2列目にはそのサンプルが属するグループを指定する。最低2つのグループが必要。
最後に正規化(ノーマライズ)の方法などのパラメータを指定できる。

使用されている正規化法についてはNORMSEQの論文の表1を確認してください。それぞれ引用もされています。
チュートリアルの説明。

最小リードカウント

Parameters
上のアノテーションファイルで定義されたグループ間の差分発現解析はデフォルトで実行される。Noを選択すると無効にできる。

差分発現解析のカットオフとするFDRの値

バッチエフェクトの補正。デフォルトではNo。

バッチ効果は、物流や実用上の制約により、あるサンプルの「バッチ」が他のサンプルと異なる方法で処理された場合に生じる(例;異なるシークエンシングで行われた実験や場所が異なる、人が異なる。ライブラリ作成プロトコルが異なる、キットのロットが異なるなど)。このようなサンプルのバッチ間の差は、実験の技術的なばらつきとなり、下流の生物学的分析に好ましくない影響を与える可能性がある。計算によるバッチ補正は、このような制約により実験的なバッチの影響を避けることができない場合に適用する(マニュアルより)。
バッチエフェクト補正を選択した場合、バッチアノテーションファイルをアップロードする。アノテーションファイルと同様に、2列のファイルで、1列目にサンプル名、2列目にそのサンプルが属するバッチエフェクトグループを記載する。

サブミットする。

出力例
結果は4つのタブ: Summary、Visualization、Differential Expression、Downloadsに整理されており、ユーザーが正規化の効果を段階的に調べることができるように設計されている。

結果は15日間保存される。
1、サマリータブ
ダウンストリーム解析に最適な正規化方法を選択する作業を支援するためのタブ。

helpなし

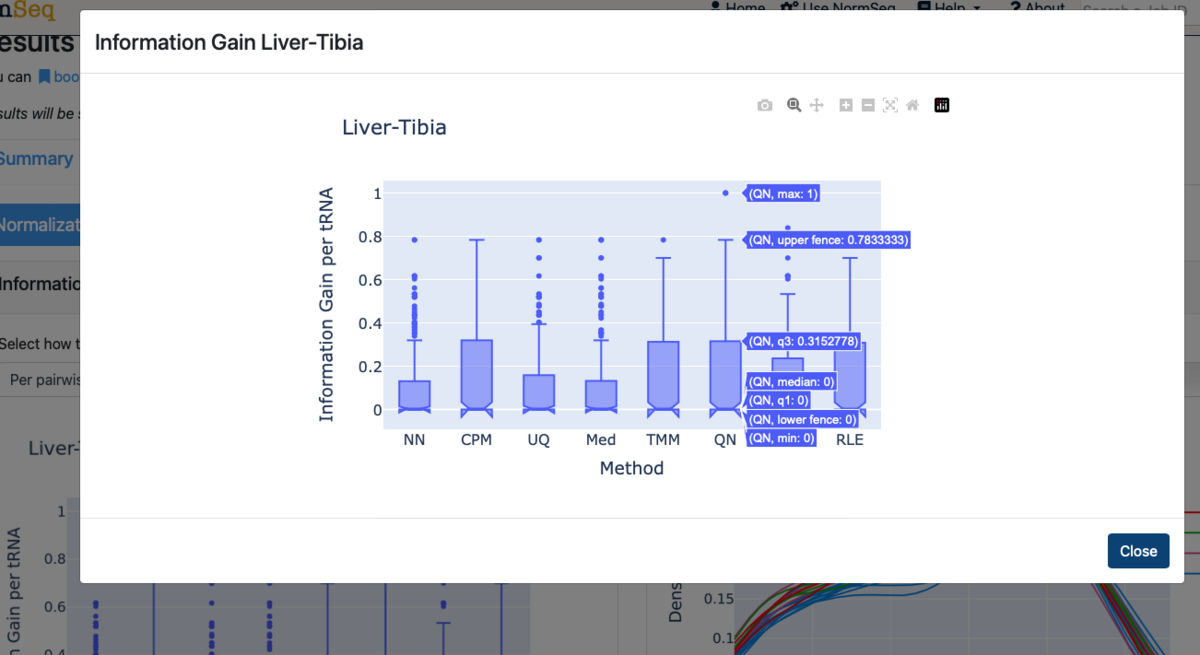
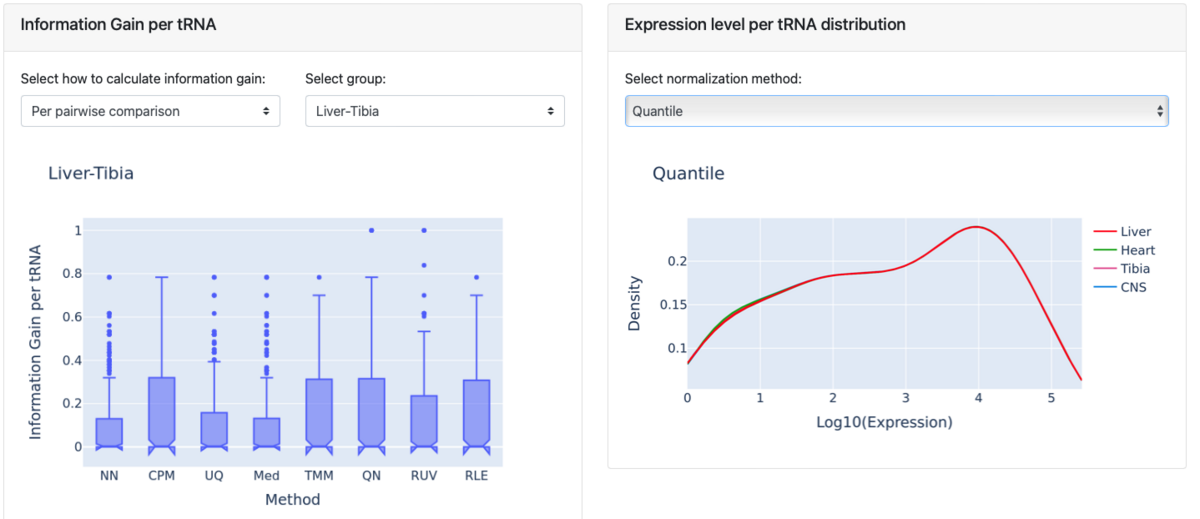
最初のプロットは、ユーザーが提出前に選択した各正規化法のノイズ低減を知らせる。これは0(ノイズが最も多い)と1(ノイズが最も少ない)の間の値を持つ。information gainの値が高いほど、そのRNAの生物学的グループに基づく明確な分離を表す。
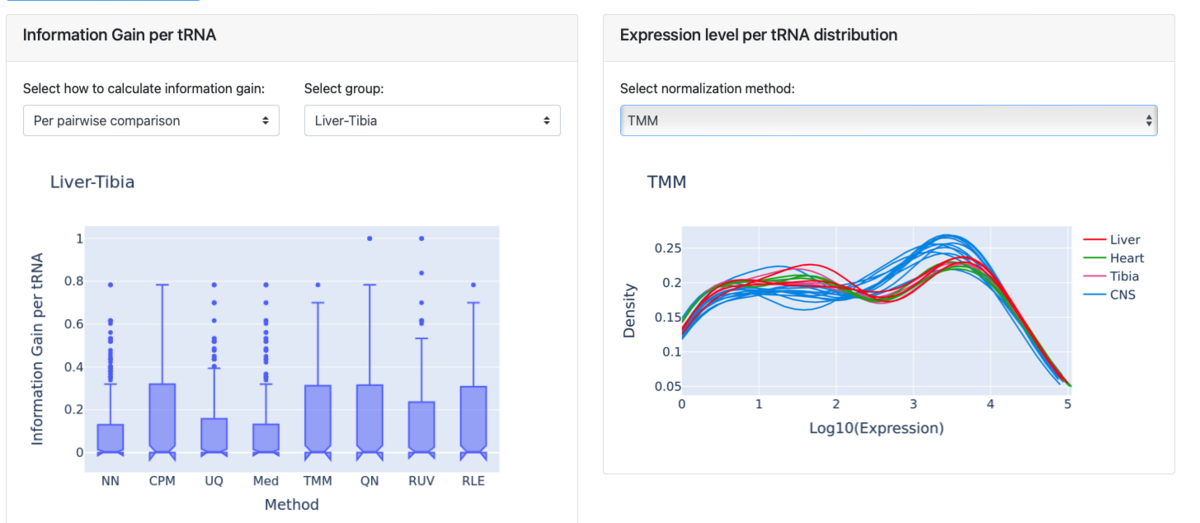
グループ毎の比較と条件間のペアワイズ比較が選べる。下図はグループ毎の正規化法の比較で、Liverグループについての結果となる。

グループごとの分布を選択した場合、より高いinformation gain値は、他のすべてのグループと比較したそのグループの差異が、その特定の方法の時、より明白であることを示す。

ペアワイズの比較が選択された場合、より高いinformation gain値を持つ正規化法がその特定の比較の生物学的シグナルをよりよく回復させると解釈できる。

図はクリックすることでインタラクティブなプロットに切り替えできる。
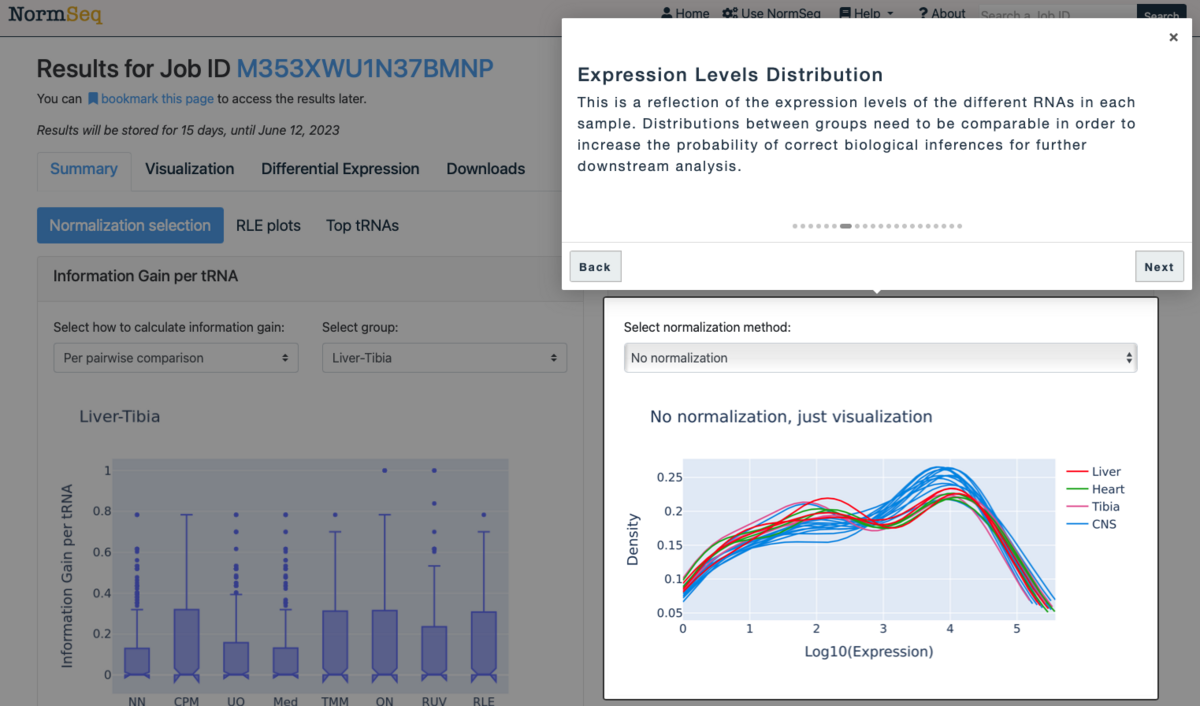
右の図は各サンプルの発現レベルの分布。グループ間の分布は、下流の分析で正しい生物学的推論を行う確率を高めるために、比較可能である必要がある。
raw => TMMに変更
raw => CPM

raw => Quantile
SummaryのTop RNAタブ
このタブでは、TOP10の存在量の10種類のRNAを、グループ間の平均発現量または倍率変化で並べた基本的なプロットが表示される。
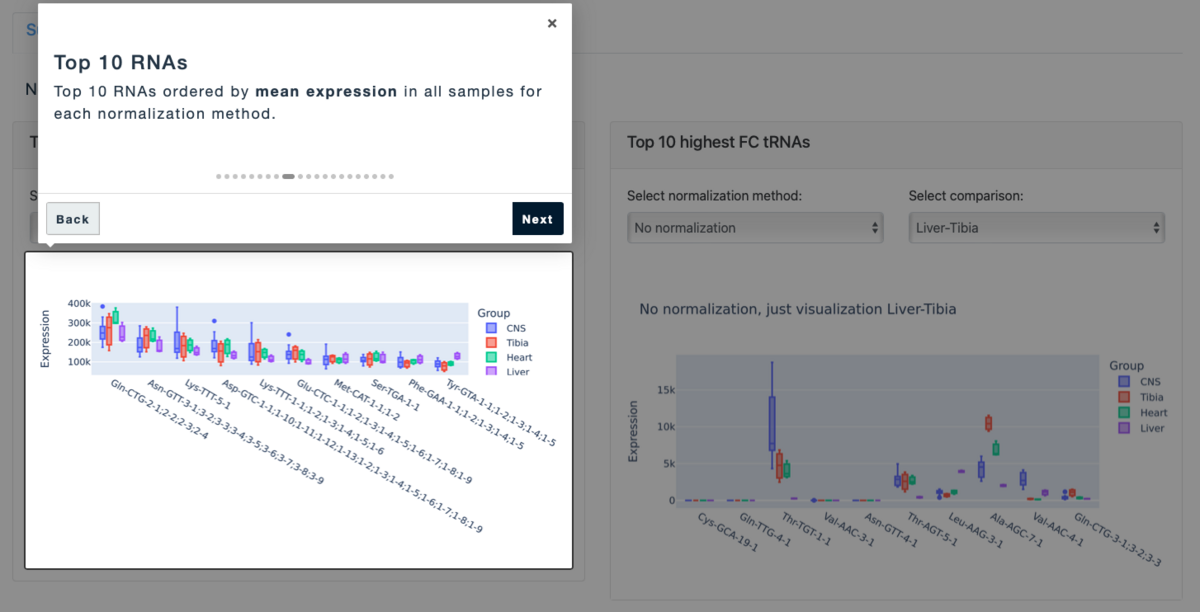
図は正規化なし。上のボタンから正規化方法を選んで図を変更できる。
上位10個のRNAをFold-Changeで並べたもので、潜在的なグループ比較と各正規化方法によるもの。
図は正規化なし。上のボタンから正規化方法を選んで図を変更できる。
2、Cisualizationタブ
視覚化のタブ。Hyerarchical clustering、PCA、Expression per RNAの3つのセクションに分かれている。
階層的クラスタリング(Hyerarchical clustering)
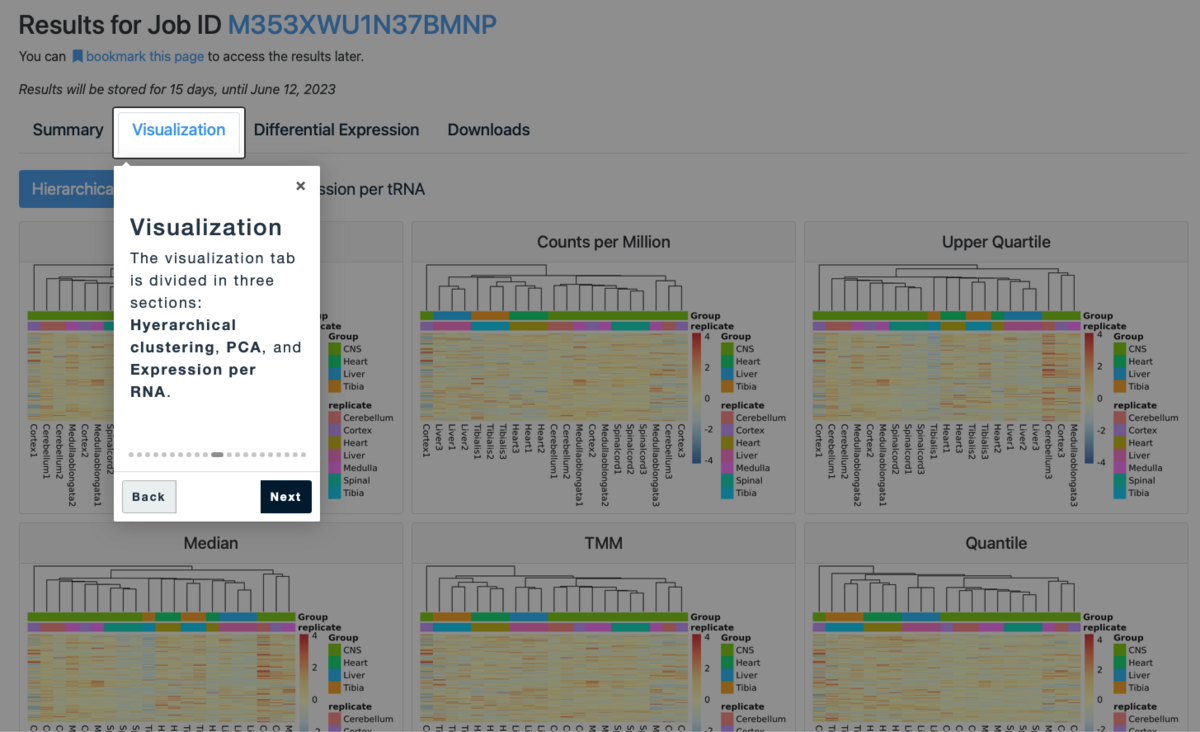
階層的クラスタリング解析は、選択した正規化方法それぞれについて、サンプル間の類似性で並べて可視化する。サンプルが所属するグループによって色分けされる。
図をクリックすると、クラスタリング手法をSingle, complete, average or medianから選べるインタラクティブな図に切り替わる。

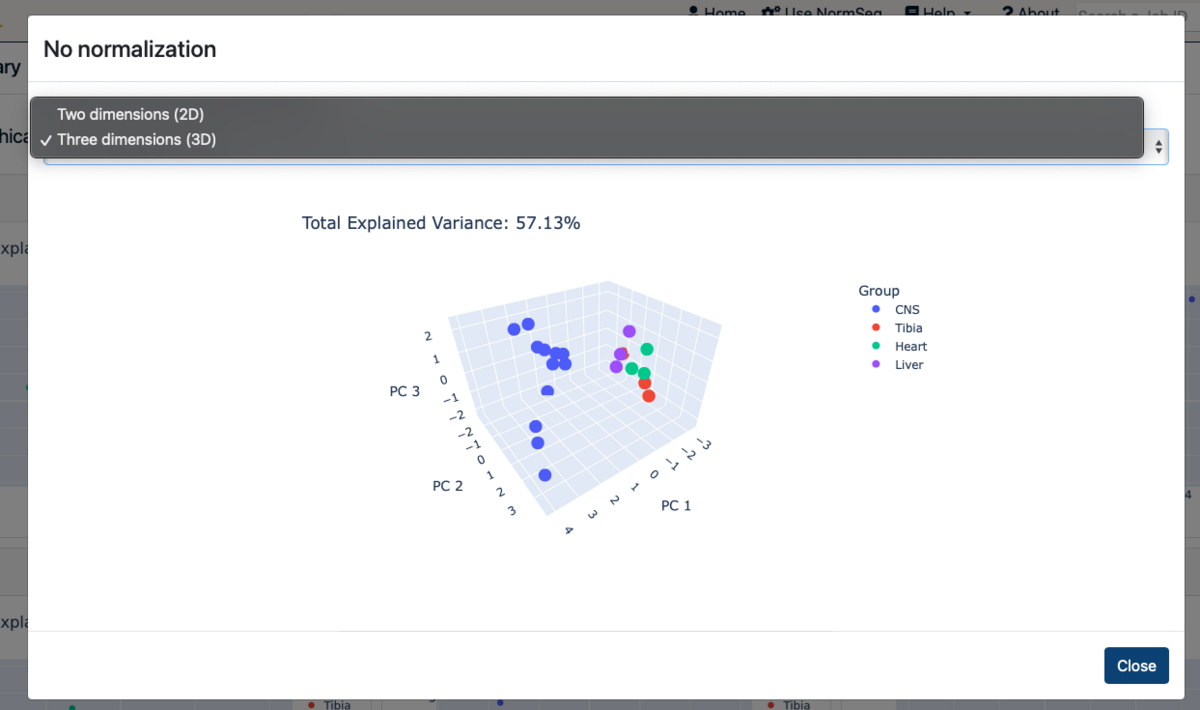
PCAタブ
分析されたすべての正規化について表示される。各グループは異なる色でプロットされる。
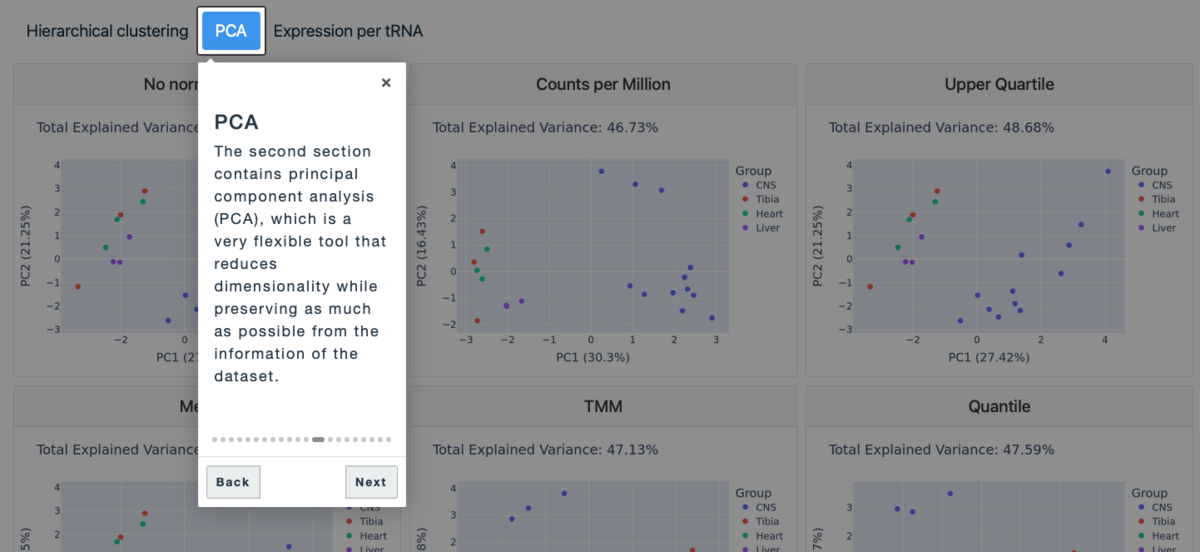
図をクリックするとインタラクティブな3次元プロットに切り替えできる。
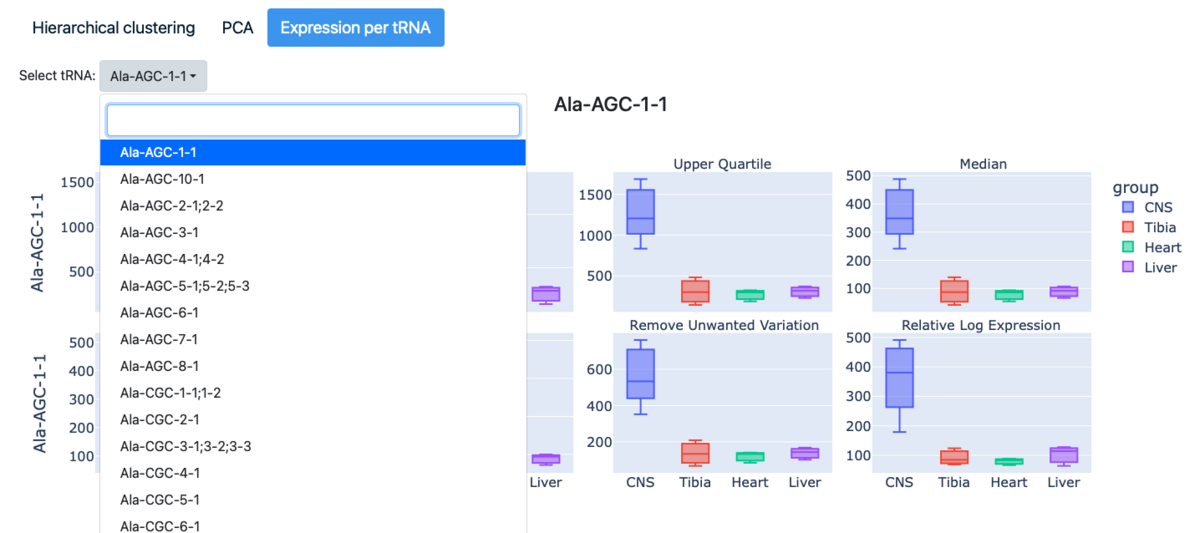
Expression per tRNA
RNA分子ごとのプロット。デフォルトでは何も表示されていない。遺伝子を選択するとプロットされる。

3、Differential Expression
Differential Expression (DE)解析のタブ。実験グループ間の発現レベルの差分解析を行う。3つのセクションに分かれている。
Summaryでは、DEの数がedgeR、NOISeq、T-testについてまとめられている(DESeq2もある)。

Results per methodでは、各差分発現解析での発現変動遺伝子のリストを確認できる。

Consensus Differential Expressionでは、edgeR、NOISeq、T-test、DESeq2間のコンセンサスを計算する。

関心のある遺伝子の変動の可能性について総合的に評価できるこのタブは特に有用と考えられる。
4、Downloads
右端のタブから、全ての結果を含む圧縮ファイルをダウンロードすることができる。

コメント
30サンプル(列数)くらいある発現マトリックスを解析してみると、30分くらいで結果のページがロードされました。
引用
NORMSEQ: a tool for evaluation, selection and visualization of RNA-Seq normalization methods
Chantal Scheepbouwer, Michael Hackenberg, Monique A J van Eijndhoven, Alan Gerber, Michiel Pegtel, Cristina Gómez-Martín
Nucleic Acids Res. 2023 May 22
関連