最近のブレイクスルーで、深層学習を用いて複数配列アラインメント(MSA)の進化情報を利用し、タンパク質の構造を正確に予測することができるようになった。しかし、オーファンタンパク質や抗体のような進化の早いタンパク質のように、相同タンパク質のMSAが常に利用できるとは限らない。また、タンパク質は通常、一次アミノ酸配列からその三次元構造へと自然環境下で折り畳まれるため、進化情報やMSAは、タンパク質の折り畳み形態の予測には必要ないはずである。ここでは、一次配列のみから高分解能のタンパク質構造を予測することに成功した初めての計算手法であるOmegaFoldを紹介する。単一配列からの予測を可能にするタンパク質言語モデルと、タンパク質構造で訓練された幾何学に着想を得た変換モデルを新たに組み合わせることで、OmegaFoldはRoseTTAFoldを上回り、最近公開された構造ではAlphaFold2と同等の予測精度を達成した。OmegaFoldは、機能的に特徴付けられたタンパク質ファミリーに属さないオーファンタンパク質や、速い進化のためにMSAがノイズになりがちな抗体について、正確な予測を可能にした。本研究は、構造予測における多くのギャップを埋め、自然界におけるタンパク質フォールディングの理解に一歩近づいた。
Protein structure can be predicted from a single sequence alone with high accuracy. @HelixonBio team have developed OmegaFold, achieving performance similar to RF and AF2's MSA versions. Only a single sequence is given as input. 1/5 pic.twitter.com/FYNP1eJDO0
— Jian Peng (@peng_illinois) June 19, 2022
(2022年のツイート)
コマンドの実行方法だけ簡単に紹介します。
インストール
mambaでpython3.10の環境を作ってテストした(ubuntu18LTS、GPU: GTX 1080)。
mamba create -n omegafold python=3.10
conda activate omegafold
#本体
git clone https://github.com/HeliXonProtein/OmegaFold
cd OmegaFold
python setup.py install
> omegafold --help
usage: omegafold [-h] [--num_cycle NUM_CYCLE] [--subbatch_size SUBBATCH_SIZE] [--device DEVICE] [--weights_file WEIGHTS_FILE] [--weights WEIGHTS] [--model MODEL] [--pseudo_msa_mask_rate PSEUDO_MSA_MASK_RATE] [--num_pseudo_msa NUM_PSEUDO_MSA]
[--allow_tf32 ALLOW_TF32]
input_file output_dir
Launch OmegaFold and perform inference on the data. Some examples (both the input and output files) are included in the Examples folder, where each folder contains the output of each available model from model1 to model3. All of the results are obtained by
issuing the general command with only model number chosen (1-3).
positional arguments:
input_file The input fasta file
output_dir The output directory to write the output pdb files. If the directory does not exist, we just create it. The output file name follows its unique identifier in the rows of the input fasta file"
options:
-h, --help show this help message and exit
--num_cycle NUM_CYCLE
The number of cycles for optimization, default to 10
--subbatch_size SUBBATCH_SIZE
The subbatching number, the smaller, the slower, the less GRAM requirements. Default is the entire length of the sequence. This one takes priority over the automatically determined one for the sequences
--device DEVICE The device on which the model will be running, default to the accelerator that we can find
--weights_file WEIGHTS_FILE
The model cache to run, default os.path.expanduser("~/.cache/omegafold_ckpt/model.pt")
--weights WEIGHTS The url to the weights of the model
--model MODEL The model number to run, current we support 1 or 2
--pseudo_msa_mask_rate PSEUDO_MSA_MASK_RATE
The masking rate for generating pseudo MSAs
--num_pseudo_msa NUM_PSEUDO_MSA
The number of pseudo MSAs
--allow_tf32 ALLOW_TF32
if allow tf32 for speed if available, default to True
実行方法
タンパク質配列、出力ディレクトリを使用する。初回はweightファイルがダウンロードされてからランされる(自分の場合は/home/kazu/.cache/omegafold_ckpt/model.pt)。
omegafold INPUT.fasta outdir

ラン中はGPU使用率が100%になる(GTX 1080 8GBメモリ)。

(nvtop使用)
試した時は1、2分で終了した(360アミノ酸長)。
outdir/
![]()
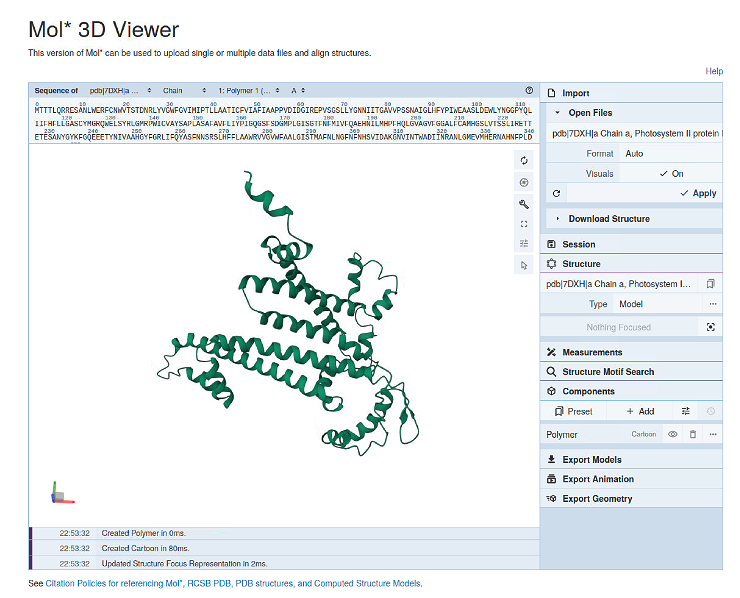
Mol* 3D ViewerでPDBファイルを開いた。
引用
High-resolution de novo structure prediction from primary sequence
Ruidong Wu, Fan Ding, Rui Wang, Rui Shen, Xiwen Zhang, Shitong Luo, Chenpeng Su, Zuofan Wu, Qi Xie, Bonnie Berger, Jianzhu Ma, Jian Peng
bioRxiv, Posted July 22, 2022
参考
OmegaFoldはMSA取得処理が必要ないので大容量SSDなどが必要ないしColabFoldよりも超爆速な代わりに、GPUメモリをたくさん食うらしいので、16GB GPUで580残基くらいまでしか予測できないらしい。
— Yoshitaka Moriwaki (@Ag_smith) August 4, 2022
残基数の短いde novoタンパク質、MSAが激レアなタンパク質、ペプチド構造予測にはかなり使えそうですね
参考になりました。ありがとうございます。