2022/09/07, 9/8 追記
微生物群は、人間の病気や生理活動に不可欠な役割を担っている。ゲノム配列の株レベルの違いにより、微生物の機能が異なることがある。ショットガン・メタゲノムシーケンスを用いると、微生物群集の菌株を実用的にプロファイリングすることができる。しかし、菌株間の配列が非常に類似しているため、現在の手法は未発達である。本著者らは、遺伝子型頻度から、同じ一塩基変異(SNV)遺伝子座における菌株の遺伝子型を推定できることを確認した。また、同じリードがカバーする異なる遺伝子座のバリアントは、同一株上に存在する証拠となる可能性がある。
PStrainは、MetaPhlAn2マーカー遺伝子のSNVに基づき、遺伝子型頻度と複数のSNV座をカバーするリードを利用して、反復的に株のプロファイリングを行う最適化手法である。PStrainは、従来の手法と比較して、菌株の存在量と遺伝子型の推定性能をそれぞれ平均87.75%と59.45%向上させることに成功した。PStrainを大腸ガンの2つのコホートのデータセットに適用したところ、Bacteroides coprocola株の配列がCRCと対照サンプルで有意に異なることがわかった。これは、CRCの腸内細菌叢におけるB.coprocolaの役割の可能性を初めて報告するものである。
インストール
依存
Third party pipelines:
- SamTools-1.3.1
- bowtie2-2.3.1
- metaphlan2/3
- GATK-3.5 (java-1.8.0_241)
- picard-tools-2.1.0
mamba create -n PStrain python=3 -y
conda activate PStrain
pip install numpy scipy pandas pulp pysam pickle5
#third party (GATkは登録が必要*1)
mamba install -c bioconda gatk==3.5 -y
mamba install -c bioconda samtools -y
mamba install -c bioconda bowtie2 -y
mamba install -c bioconda picard -y
#本体
git clone https://github.com/wshuai294/PStrain.git
cd PStrain/scripts/
> python3 PStrain.py -h
usage: python3 PStrain.py [options] -c/--conf <config file> -o/--outdir <output directory>
The config file should follow the format:
//
sample: [sample1_ID]
fq1: [forward reads fastq]
fq2: [reverse/mate reads fastq]
//
sample: [sample2_ID]
fq1: [forward reads fastq]
fq2: [reverse/mate reads fastq]
...
Help information can be found by python3 PStrain.py -h/--help, config file format for single end reads , and additional information can be found in README.MD or https://github.com/wshuai294/PStrain.
PStrain: profile strains in shotgun metagenomic sequencing reads.
required arguments:
-c , --conf The configuration file of the input samples.
-o , --outdir The output directory.
options:
-h, --help show this help message and exit
-p , --proc The number of process to use for parallelizing the whole pipeline (default is 1), run a sample in each process.
-n , --nproc The number of CPUs to use for parallelizing the mapping with bowtie2(default is 1).
-w , --weight The weight of genotype frequencies while computing loss, then the weight of linked read type frequencies is 1-w. The value is between 0~1. (default is 0.0)
--lambda1 The weight of prior knowledge while rectifying genotype frequencies. The value is between 0~1. (default is 0.0)
--lambda2 The weight of prior estimation while rectifying second order genotype frequencies. The value is between 0~1. (default is 0.0)
--species_dp The minimum depth of species to be considered in strain profiling step (default is 5).
--snp_ratio The SNVs of which the depth are less than the specific ratio of the species's mean depth would be removed. (default is 0.45).
--qual The minimum quality score of SNVs to be considered in strain profiling step (default is 20).
--similarity The similarity cutoff of hierachical clustering in merge step (default is 0.95).
--elbow The cutoff of elbow method while identifying strains number. If the loss reduction ratio is less than the cutoff, then the strains number is determined. Default is 0.24.
--bowtie2 Path to bowtie2 binary. If your platform is not Linux, you need to specify your own bowtie2 binary.
--bowtie2-build Path to bowtie2 binary. If your platform is not Linux, you need to specify your own bowtie2-build binary.
--samtools Path to samtools binary (default version is in the packages path). If your platform is not Linux, you need to specify your own samtools binary.
--metaphlan2 Path to metaphlan2 script (default version is in the packages path).
--gatk Path to gatk binary (default version is in the packages path).
--picard Path to picard binary (default version is in the packages path).
--dbdir Path to marker gene database (default is in the ../db path).
--prior The file of prior knowledge of genotype frequencies in the population.
> python3 PStrain_V30.py -h
usage: python3 PStrain.py [options] -c/--conf <config file> -o/--outdir <output directory>
The config file should follow the format:
//
sample: [sample1_ID]
fq1: [forward reads fastq]
fq2: [reverse/mate reads fastq]
//
sample: [sample2_ID]
fq1: [forward reads fastq]
fq2: [reverse/mate reads fastq]
...
Help information can be found by python3 PStrain.py -h/--help, config file format for single end reads , and additional information can be found in README.MD or https://github.com/wshuai294/PStrain.
PStrain: profile strains in shotgun metagenomic sequencing reads.
required arguments:
-c , --conf The configuration file of the input samples.
-o , --outdir The output directory.
optional arguments:
-h, --help show this help message and exit
-p , --proc The number of process to use for parallelizing the
whole pipeline (default is 1), run a sample in each
process.
-n , --nproc The number of CPUs to use for parallelizing the
mapping with bowtie2(default is 1).
-w , --weight The weight of genotype frequencies while computing
loss, then the weight of linked read type frequencies
is 1-w. The value is between 0~1. (default is 0.0)
--lambda1 The weight of prior knowledge while rectifying
genotype frequencies. The value is between 0~1.
(default is 0.0)
--lambda2 The weight of prior estimation while rectifying second
order genotype frequencies. The value is between 0~1.
(default is 0.0)
--species_dp The minimum depth of species to be considered in
strain profiling step (default is 5).
--snp_ratio The SNVs of which the depth are less than the specific
ratio of the species's mean depth would be removed.
(default is 0.45).
--qual The minimum quality score of SNVs to be considered in
strain profiling step (default is 20).
--similarity The similarity cutoff of hierachical clustering in
merge step (default is 0.95).
--elbow The cutoff of elbow method while identifying strains
number. If the loss reduction ratio is less than the
cutoff, then the strains number is determined. Default
is 0.24.
--bowtie2 Path to bowtie2 binary. If your platform is not Linux,
you need to specify your own bowtie2 binary.
--bowtie2-build Path to bowtie2 binary. If your platform is not Linux,
you need to specify your own bowtie2-build binary.
--samtools Path to samtools binary (default version is in the
packages path). If your platform is not Linux, you
need to specify your own samtools binary.
--metaphlan3 Path to metaphlan3 (default version is in the packages
path).
--metaphlan3_output_files
If you have run metaphlan3 already, give metaphlan3
result file in each line, the order should be the same
with config file. In particular, '--tax_lev s' should
be added while running metaphlan3.
--gatk Path to gatk binary (default version is in the
packages path).
--picard Path to picard binary (default version is in the
packages path).
--dbdir_V30 Path to marker gene database (default is in the
../db_V30 path).
--prior The file of prior knowledge of genotype frequencies in
the population. Not providing this is also ok.
実行方法
ランするにはfastqのパスとサンプル名の関係を示したconfigファイルが必要。
https://github.com/wshuai294/PStrain/blob/master/test/config.txt
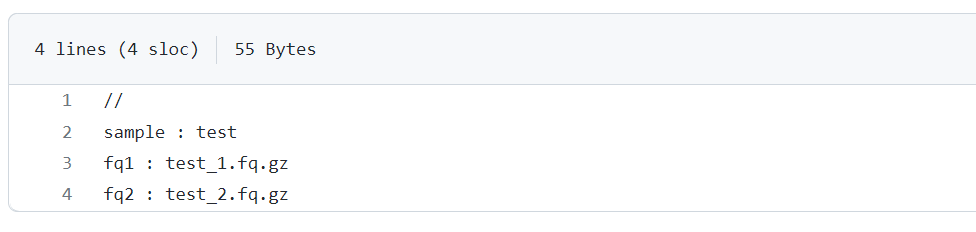
マーカー遺伝子のDBはオーサーによって準備されている(リンク)。ダウンロードしてディレクトリに配置する。
DB_dir/

configファイル、CPU数、出力ディレクトリ、マーカーDB、サードパーティのツールのパスを指定して実行する。
python3 PStrain_V30.py -c config.txt -n 30 -o out --metaphlan3 <path>/<to>/metaphlan --bowtie2 <path>/<to>/bowtie2 --bowtie2-build <path>/<to>/bowtie2-build --samtools <path>/<to>/samtools --picard <path>/<to>/picard.jar --gatk <path>/<to>/GenomeAnalysisTK.jar --dbdir_V30 <path>/<to>/<DB_dir>
- -n The number of CPUs to use for parallelizing the mapping with bowtie2(default is 1).
- -o The output directory
- -c The configuration file of the input samples.
出力
"-o"で指定した出力ディレクトリには、configファイルで指定されたsample IDそれぞれのサブディレクトリ、全サンプルをマージしたmergeというサブディレクトリができる。
sample IDのサブディレクトリの例

サブディレクトリ中のresultに結果が保存される。

strain_RA.txtにはどの種にどれだけの株が見つかったのか示されている。
strain_RA.txtの例(Githubより)
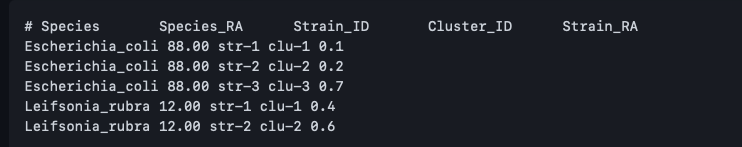
2列目のSpecies_RA カラムは、サンプル中の種の相対的な存在量、3列目のStrain_ID 列は、サンプル中の各生物種の菌株ID、4列目のCluster_ID カラムは、全サンプル中の菌株の対応するクラスタを意味する。
- mergeというサブディレクトリには、どの種にどれだけの株が見つかったのかの個数がまとめられてたファイル、全サンプル中の各菌株クラスターのコンセンサス配列、全サンプル中の各生物種の株番号が示されたファイル、その他が出力される。
- 自分で簡単に種固有のパイプラインを構築できる(Gihtub参照)。
- PStrain-tracerというPStrainの下流解析のパイプラインも開発されている。PStrain-tracerは、全ゲノムショットガンメタゲノミクスデータから、ゲノム配列や株の性質を推定するツールで、Fecal microbiota transplantation (FMT) の分析に使用されている(論文)。
- PStrain-tracerにはテストスクリプトが付属しており、簡単に動作確認できる(依存関係を揃えて試したところ、問題なく全結果が出力された)。
引用
PStrain: an iterative microbial strains profiling algorithm for shotgun metagenomic sequencing data
Shuai Wang, Yiqi Jiang, Shuaicheng Li Author Notes
Bioinformatics, Volume 36, Issue 22-23, 1 December 2020, Pages 5499–5506
関連
*1
GATK3のダウンロード
ダウンロード後、gatk-registerをバイナリを指定して叩くとコピーされパスが通る。
#初回はgatk-registerを走らせて実行ファイルのパスを叩く
gatk-register <path>/<to>/<your>/GenomeAnalysisTK.jar
その後、PStrainを実行するときはJava Archive(.JARファイル)の方を指定する。Picardも同様。