ナノポアシーケンスは長いリードを生成し、特にドラフト細菌ゲノムのアセンブリにおいて、次世代シーケンシングと比較してユニークな利点を提供する。しかし、データの特性やアセンブリアルゴリズムに起因するアセンブリエラーが発生することがある。これらの問題を解決するために、本著者は同一のナノポアシーケンスデータの複数のアセンブリからコンセンサス配列を生成し、エラー修正を行うパイプライン、MAECIを開発した。系統的な評価により、MAECIは細菌ゲノムアセンブリの精度と完全性を向上させる効率的かつ効果的なパイプラインであることが示された。利用可能なコードと実装は https://github.com/langjidong/MAECI にある。
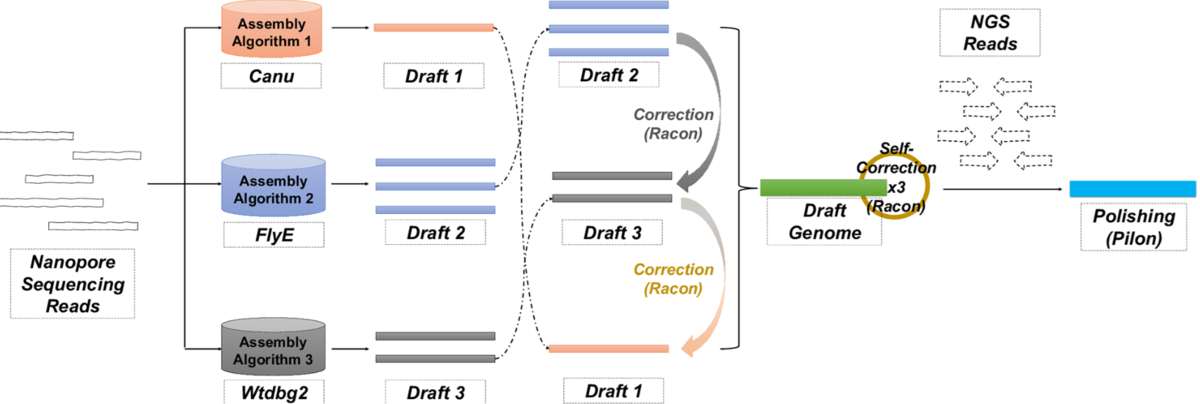
Overview of the MAECI assembly pipeline. 論文より転載
インストール
説明がないので、perlスクリプトの内容を見て必要なツールを導入した。
mamba create -n maeci -y
conda activate maeci
mamba install -y -c bioconda wtdbg racon canu flye porechop bwa samtools pilon samtools sambamba nanoplot quast
git clone https://github.com/langjidong/MAECI.git
cd MAECI/
> perl Multiple-Assembly-Integration.pl
===============================================
Edit by Jidong Lang; E-mail: langjidong@hotmail.com;
===============================================
Option
-fq <Input File> Input *.fq file
-path1 <Assembly Method 1> The path of assembly method/software, default: /usr/bin/canu
-path2 <Assembly Method 2> The path of assembly method/software, default: /usr/bin/flye
-path3 <Assembly Method 3> The path of assembly method/software, default: /usr/bin/wtdbg2
-path4 <Correction Method> The path of self-correction method/software, default: /usr/bin/racon
-genome_size <Estimated Genome Size> Estimated genome size, the unit is mb
-ngs_polishing <yes:1|no:0> Whether need NGS data for polishing, default: 0
-ngs_file1 <NGS Fastq file1> The ngs fastq file, such as read_1.fq.gz
-ngs_file2 <NGS Fastq file2> The ngs fastq file, such as read_2.fq.gz
-outputdir <Output Dir> The output results pathdir
-reference <Reference Genome> The reference genome of similar species
-process <Number of process used> N processes to use, default is 1
-help print HELP message
Example:
perl Multiple-Assembly-Integration.pl -fq nanopore.fq -path1 canu -path2 flye -path3 wtdbg2 -path4 racon -genome_size 1m -ngs_polishing 1 -ngs_file1 read_1.fq.gz -ngs_file2 read_2.fq.gz -outputdir ./outputdir -reference reference.fasta -process 8
Note: There are other options for assembling methods/softwares, and theoretically one method is also possible. But we suggest that should better choose at least 3 methods. If you want to change the assembly methods, please modify the command line (Line 75-87) in this script.
実行方法
3つ以上のアセンブラの使用が推奨されている。ロングリードとアセンブラのパスを指定する。
perl ../Multiple-Assembly-Integration.pl -fq test.fq -path1 canu -path2 flye -path3 wtdbg2 -path4 racon -genome_size 4.5m -ngs_polishing 1 -outputdir
./ -reference /GCF_000005845.2_ASM584v2_genomic.fna -ngs_file1 /NGS/test_1.fq.gz -ngs_file2 /NGS/test_2.fq.gz -process 16 > log 2>&1
root直下に作業ディレクトリを作ろうとするなど、気になる点があります。使用される場合は注意して下さい。
引用
MAECI: A pipeline for generating consensus sequence with nanopore sequencing long-read assembly and error correction
Jidong Lang
PLoS One. 2022 May 20;17(5):e0267066
関連
参考