ChIP-sequencing、RNA-sequencing、DNA sequencing、定量的メタボロミクスなどのハイスループット技術により、膨大な量のデータが生成される。研究者は、これらのハイスループット研究から影響を受けた遺伝子の生物学的意義を解釈するために、しばしばfunctional enrichment toolsに依存している。しかし、現在利用可能なfunctional enrichment toolsは、機能データベースリポジトリからの新しいエントリに適応するために頻繁に更新される必要がある。そこで、KEGG、Reactome、Gene Ontology などのソースデータベースから直接更新された情報を使用して、機能エンリッチメント解析を実行できる簡便なツールが必要とされている。
本研究では、GeneSCF (Gene Set Clustering based on Functional annotations) と呼ばれるコマンドラインツールを設計し、遺伝子セットの機能関連情報をリアルタイムで予測することに焦点を当てた。このツールは、KEGG, Reactome, Gene Ontologyなどの著名な機能データベースから、4000以上の生物の情報を扱うことができるように設計されている。本ツールを2つの公開データセットに適用し、生物学的に関連する機能情報を予測することに成功した。
GeneSCFは、リアルタイムに参照機能データベースを用いてエンリッチメント解析を行うことができるため、他のエンリッチメントツールと比較して信頼性が高いと言える。また、ハイスループット・データの下流解析に利用できる他のパイプラインとの統合が容易なツールである。さらに重要なことは、GeneSCFは複数の遺伝子リストを異なる生物種で同時に実行することができるため、ユーザーの時間を節約することができる。このツールはすぐに使えるように設計されているため、複雑なコンパイルやインストール手順は必要ない。
特徴
- 多くの生物種に対応。
- 複数のソースデータベースを用いて、複数の遺伝子リストのエンリッチメントを一度に解析することが可能(GO、KEGGのみ対応)。
- GOの語彙/パスウェイ/ファンクションと関連する遺伝子をシンプルな表形式でプレーンテキストファイルでダウンロードできる。
インストール
ubuntu18でテストした。
依存
GeneSCFはLinuxシステムでのみ動作し、Ubuntu, Mint, Cent OSで正常に動作することが確認されている。他のLinuxディストリビューションでも動作する可能性はある。
For graphical output or plots
- R (version > 3.0) and 'ggplot2' is required
git clone https://github.com/genescf/GeneSCF.git
cd GeneSCF/
export PATH=$PATH:$PWD
> ./geneSCF -h
GeneSCF USAGE:
./geneSCF -m=[update|normal] -i=[INPUT_PATH/INPUT_FILE] -t=[gid|sym] -o=[OUTPUT_PATH/FOLDER/] -db=[GO_all|GO_BP|GO_MF|GO_CC|KEGG|REACTOME|NCG] -p=[yes|no] -bg=[#TotalGenes] -org=[see,org_codes_help]
==========
Options: ALL PARAMETERS MUST BE SPECIFIED
==========
[-m= | --mode=] For normal mode use 'normal' and for update mode use 'update' without quotes.
[-i= | --infile=] Input file contains list of Entrez GeneIDs or OFFICIAL GENE SYMBOLS.
The genes must be new lines seperated (One gene per line).
[-t= | --gtype=] Type of input in the provided list either Entrez GeneIDs 'gid'
or OFFICIAL GENE SYMBOLS 'sym' (Without quotes, Example for
human 'sym' => HUGO gene symbols).
[-db= | --database=] Database you want to find gene enrichment which is either
geneontology 'GO_all' or geneontology-biological_process
'GO_BP' or geneontology-molecular_function 'GO_MF' or
geneontology-cellular_components 'GO_CC' or kegg 'KEGG' or
reactome 'REACTOME' or Network of Cancer Genes 'NCG' (Without quotes).
[-o= | --outpath=] Existing directory to save output file. The output will be with saved in the
provided location as {INPUT_FILE_NAME}_{database}_functional_classification.tsv
(tab-seperated file).
[-bg= | --background=] Total background genes to consider (Example : ~20,000 for human).
[-org= | --organism=] Please see organism codes(For human in KEGG ->'hsa' in Geneontology -> 'goa_human').
For REACTOME and NCG use 'Hs' (human).
[-p= | --plot=] For additional graphical output use 'yes' or 'no'.This requires R version > 3.0 and
'ggplot2' R package to be pre-installed on the system.
[-h | --help] For displaying this help page.
prepare_database USAGE:
./prepare_database -db=[GO_all|GO_BP|GO_MF|GO_CC|KEGG|REACTOME|NCG] -org=[see,org_codes_help]
==========
Options: ALL PARAMETERS MUST BE SPECIFIED
==========
[-db= | --database=] Options:[GO_all|GO_BP|GO_MF|GO_CC|KEGG|REACTOME]
[-org= | --organism=] Options:[see,org_codes_help]
> ./prepare_database -h
prepare_database USAGE:
./prepare_database -db=[GO_all|GO_BP|GO_MF|GO_CC|KEGG|REACTOME|NCG] -org=[see,org_codes_help]
==========
Options: ALL PARAMETERS MUST BE SPECIFIED
==========
[-db= | --database=] Options:[GO_all|GO_BP|GO_MF|GO_CC|KEGG|REACTOME]
[-org= | --organism=] Options:[see,org_codes_help]
データベースの準備
デフォルトでヒト用の遺伝子オントロジー、KEGG、Reactome、NCGからなるデータベースが用意されているが、ほかのモデル生物だと用意する必要がある。そのためのコマンドが準備されている。
#GO
prepare_database -db=GO_all -org=goa_human
#KEGG
prepare_database -db=KEGG -org=hsa
#REACTOME
prepare_database -db=REACTOME -org=Hs
#NCG (catalogue of known and candidate cancer genes)
prepare_database -db=NCG -org=Hs
- -db=GO_all | GO_BP | GO_CC | GO_MF | KEGG | REACTOME | NCG
- -org=<see organism codes>
コマンドを実行すると、指定したデータベースがダウンロードされる(GeneSCF/class/lib/db/)。

-p=yesのオプションは機能しなかった。マニュアルの間違い?
実行方法
遺伝子リストに対してエンリッチメント解析を行う。
必要な遺伝子リストは、公式遺伝子シンボル(例:ヒトのHGNC、--gtype=sym)またはEntrez GeneID(--gtype=gid)形式の遺伝子リストを含むプレーンテキスト。入力遺伝子リストは、発現変動遺伝子だったりエピジェネティック修飾に異常がある遺伝子などの個々の研究において影響を受けると予測される遺伝子リストとなる。このほか、ランには、-mで指定するupdateモードとノーマルモード(-modeパラメータ)のどちらかを指定する必要がある。updateモードでは、対応するデータベースと生物からリアルタイムに機能情報を取得してエンリッチメント解析を行う。通常モードでは、updateモードにより過去に取得・利用した機能データベースに対してエンリッチメント解析を行う。
#テストラン
cd GeneSCF/
mkdir outdir1
geneSCF -m=update -i=test/H0.list -o=outdir1/ -t=sym -db=GO_all -bg=20000 --plot=yes -org=goa_human
- -m=normal | update For normal mode use normal and for update mode use *update* without quotes
- -i= <INPUT-TEXT-FILE> Input file contains list of Entrez GeneIDs or OFFICIAL GENE SYMBOLS. The genes must be new lines seperated (One gene per line)
- -t=gid | sym Type of input in the provided list either Entrez GeneIDs gid or OFFICIAL GENE SYMBOLS sym (default: *gid*)
- -db=GO_all | GO_BP | GO_CC | GO_MF | KEGG | REACTOME | NCG
- -o= <OUTPUT-DIRECTORY> Existing directory to save output file (Don't forget to use trailing slash at end of the directory name). The output will be saved in the provided location as {INPUT_FILE_NAME}_{database}_functional_classification.tsv (tab-seperated file).
- -bg= <Total background> Total number of genes to be considered as background (Example : ~20,000 for human). It is important to choose the background appropriately. Sometimes your samples do not express all the genes. For example, if you are using differentially expressed genes for gene set enrichment analysis, you must choose total number of genes detected in your experiment including control and treatment samples irrespective of their differential status as your background (NOT the total number of genes in the annotation of the corresponding organism you are working with). All the genes from the annotation can be used when working with genes found in genome-wide studies (example, ChIP-seq, WGBS, etc.,).
- -org=<see organism codes>
- -p=yes|no For additional graphical output use yes or no.This requires R version > 3.0 and ggplot2 R package to be pre-installed on the system
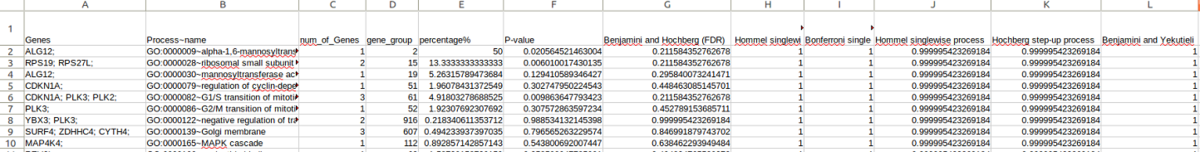
GeneSCF は、分割表(論文図1C)から、PERL の統計モジュールを利用し、標準的な多重検定補正法を用いた Fisher's Exact 検定を用いて、エンリッチメントの有意性を計算する。多重仮説補正法には、Benjamini and Hochberg、Hommel一段階処理、Bonferroni一段階処理、Hochbergステップアップ処理、Benjamini and Yekutieli多重仮説補正係数がある。GeneSCF の出力は、ユーザが提供した遺伝子リストからのヒット数に基づいてランク付けされた関数のリストがタブ区切りのテーブルになったもの。また、分割表を用いたフィッシャーの正確検定によるp値と、異なる多重仮説補正法によるFDR)値の列が含まれる(論文より)。
出力例
H0.list_GO_all_goa_human_functional_classification.tsv
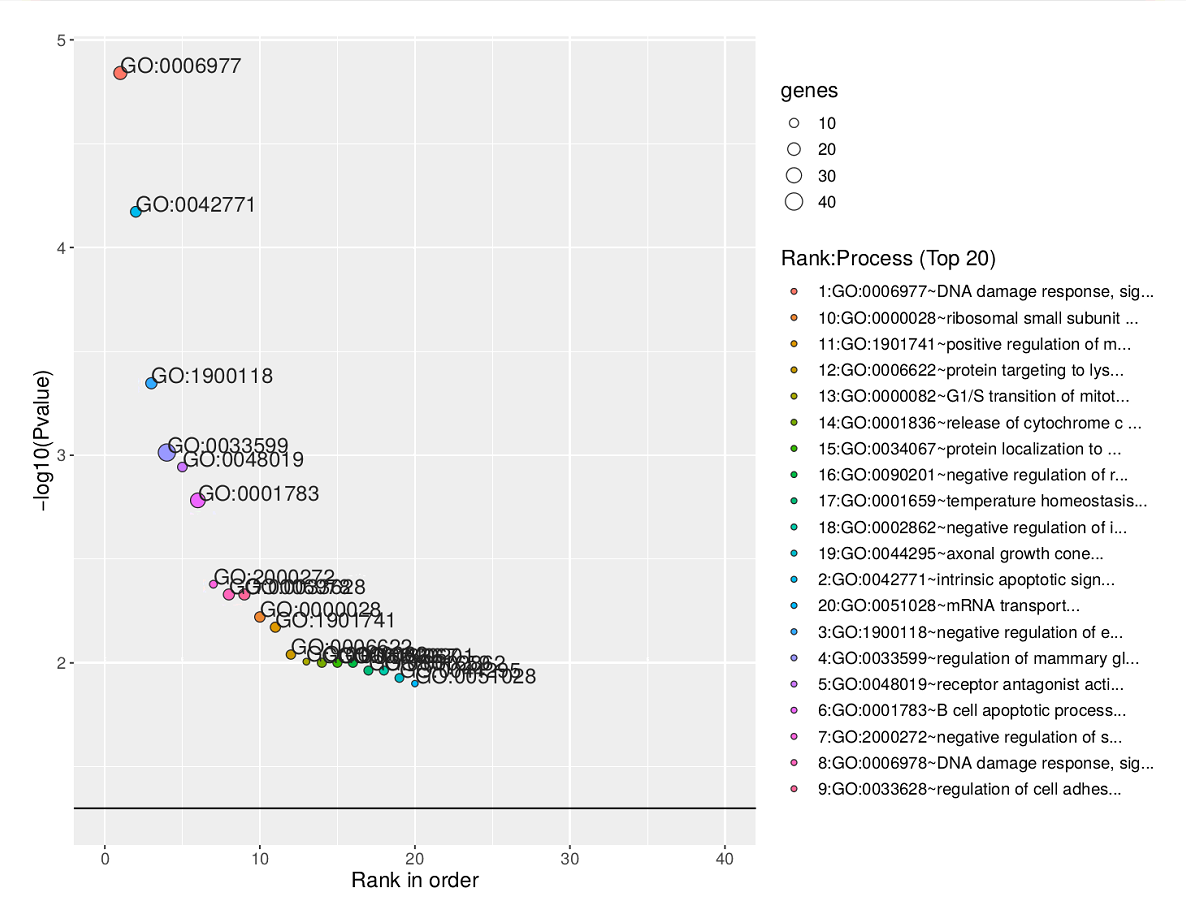
H0.list_GO_all_goa_human_enrichment_plot.pdf
mkdir outdir2
mkdir -p mapping/DB/
geneSCF -m=normal -i=test/H0.list -o=outdir2/ -t=sym -db=KEGG -bg=20000 --plot=yes -org=hsa
出力例

Githubではバッチモードの使い方についての説明もあります。アクセスして確認して下さい。
引用
GeneSCF: a real-time based functional enrichment tool with support for multiple organisms
Santhilal Subhash & Chandrasekhar Kanduri
BMC Bioinformatics volume 17, Article number: 365 (2016)