TrinotateとGOseq、Trinityのスクリプトを組み合わせることで、遺伝子セット間の機能的エンリッチメント解析を行うことができる。Trinityのマニュアルに習い、使い方を確認しておく。
インストール
ubuntu18.04でtrinityの仮想環境を作ってテストした。Rのバージョンは4.1.1。
依存
- R
- edgeR, limma, DESeq2, ctc, Biobase, gplots, ape, argparse
mamba create -n trinity python=3.8
conda activate trinity
mamba install -c bioconda -y trinity
#Trinotate
git clone https://github.com/Trinotate/Trinotate.git
#GOSeq
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("goseq")
#qvalue
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("qvalue")
> Trinotate/util/extract_GO_assignments_from_Trinotate_xls.pl
#######################################################################
#
#
# Required:
#
# --Trinotate_xls <string> Trinotate.xls file.
#
# --gene|G or --trans|T gene or transcript-mode
#
# Optional:
#
# --include_ancestral_terms|I climbs the GO DAG, and incorporates
# all parent terms for an assignment.
#
######################################################################
> trinityrnaseq/Analysis/DifferentialExpression/run_GOseq.pl
###############################################################################################
#
# --factor_labeling <string> tab delimited file with format: factor<tab>feature_id
# or
# --genes_single_factor <string> list of genes to test (can be a matrix, only the first column is used for gene IDs)
#
# --GO_assignments <string> extracted GO assignments with format: feature_id <tab> GO:000001,GO:00002,...
#
# --lengths <string> feature lengths file with format: feature_id <tab> length
#
# --background <string> gene ids file that defines the full population of genes to consider in testing.
# Ideally, these represent the genes that are expressed and relevant to this test
# as opposed to using all genes in the genome.
#
###############################################################################################
>trinityrnaseq/util/misc/TPM_weighted_gene_length.py -h
usage: TPM_weighted_gene_length.py [-h] --gene_trans_map GENE_TRANS_MAP_FILE
--trans_lengths TRANS_LENGTHS_FILE
--TPM_matrix TPM_MATRIX_FILE [--debug]
estimates gene length as isoform lengths weighted by TPM expression values
optional arguments:
-h, --help show this help message and exit
--gene_trans_map GENE_TRANS_MAP_FILE
gene-to-transcript mapping file, format:
gene_id(tab)transcript_id
--trans_lengths TRANS_LENGTHS_FILE
transcript length file, format: trans_id(tab)length
TPM expression matrix
--debug debug mode
実行方法
1、Trinotateの実行
trinotate.xlsを得る。
この後のステップ2-4は、ランに必要なファイルの準備になる。
2、GO DAG内のすべてのGOアサインの抽出
perl TRINOTATE/util/extract_GO_assignments_from_Trinotate_xls.pl \
--Trinotate_xls trinotate.xls \
--gene --include_ancestral_terms > go_annotations.txt
- --gene or --trans gene or transcript-mode
- --include_ancestral_terms climbs the GO DAG, and incorporates all parent terms for an assignment.
出力
go_annotations.txt

3、GOseqのランに必要なfactor_labeling.txtの用意 (参考ページ1, 2)
遺伝子サブセットの名称<tab>遺伝子のリストを用意する。
diff_expressed_cond_X_Y (tab) my_gene_A
diff_expressed_cond_X_Y (tab) my_gene_B
...
diff_cond_W_Z (tab) my_gene_M
diff_cond_W_Z (tab) my_gene_N
遺伝子はDEGsなどからの遺伝子ID。遺伝子サブセットの名称はユーザーが自由に決めるるが、DEGと対応した名前にする。このテキストファイルをfactor_labeling.txtという名前で保存する。
4、GOseqのランに必要なgene.lengths.txtの用意
#fasta_seq_length.plで配列の長さを取り出す。
perl trinityrnaseq/util/misc/fasta_seq_length.pl Trinity.fasta > Trinity.fasta.seq_lens
#TPM_weighted_gene_lengthのラン。DEGsのリストから配列の長さファイルを出力。各ファイルはDEG解析プロセス中で生成されているはず。
perl trinityrnaseq/util/misc/TPM_weighted_gene_length.py \
--gene_trans_map Trinity.fasta.gene_trans_map \
--trans_lengths Trinity.fasta.seq_lens \
--TPM_matrix isoforms.TMM.EXPR.matrix > Trinity.gene_lengths.txt
Trinity.gene_lengths.txtができる。これを次の5で使う。
5、GOseqのラン(機能的エンリッチメント解析)
2-4で用意したファイルを指定する。backgroundの遺伝子リストも指定する。
run_GOseq.pl --factor_labeling factor_labeling.txt \
--GO_assignments go_annotations.txt \
--lengths gene.lengths.txt --background gene_list
- --factor_labeling tab delimited file with format: factor<tab>feature_id
- --GO_assignments extracted GO assignments with format: feature_id
- --lengths feature lengths file with format: feature_id <tab> length
- --background gene ids file that defines the full population of genes to consider in testing. Ideally, these represent the genes that are expressed and relevant to this test as opposed to using all genes in the genome.
出力例
補足
go_annotations.txtとTrinity.gene_lengths.txtは発現変動遺伝子の解析時に指定して、発現変動遺伝子の計算と同時にGO enrichment解析を行うこともできる。それには、先日紹介したanalyze_diff_expr.plのランまでは同じ。それから、analyze_diff_expr.plのランで出来る出力ディレクトリに移動する。
それから前回とはオプションを変えてanalyze_diff_expr.plを走らせる。今回はgo_annotations.txtとTrinity.gene_lengths.txtのファイルを指定する。
run_DE_analysis.pl --matrix counts.matrix --samples_file list --method DESeq2 --dispersion 0.1
cd DESeq2xxx/
analyze_diff_expr.pl --matrix group_A_vs_group_B.DESeq2.count_matrix -P 1e-3 -C 2 --examine_GO_enrichment --GO_annots go_annotations.txt --gene_lengths Trinity.gene_lengths.txt
- --examine_GO_enrichment run GO enrichment analysis
- --GO_annots <string> GO annotations file
- --gene_lengths <string> lengths of genes file
- --include_GOplot optional: will generate inputs to GOplot and attempt to make a preliminary pdf plot/report for it.
有意なFDRとfold-changesを持つ転写産物と抽出し、サンプル間の発現差のパターンに従った転写物をクラスタリング、GO enrichment解析が全て実行される。
出力例
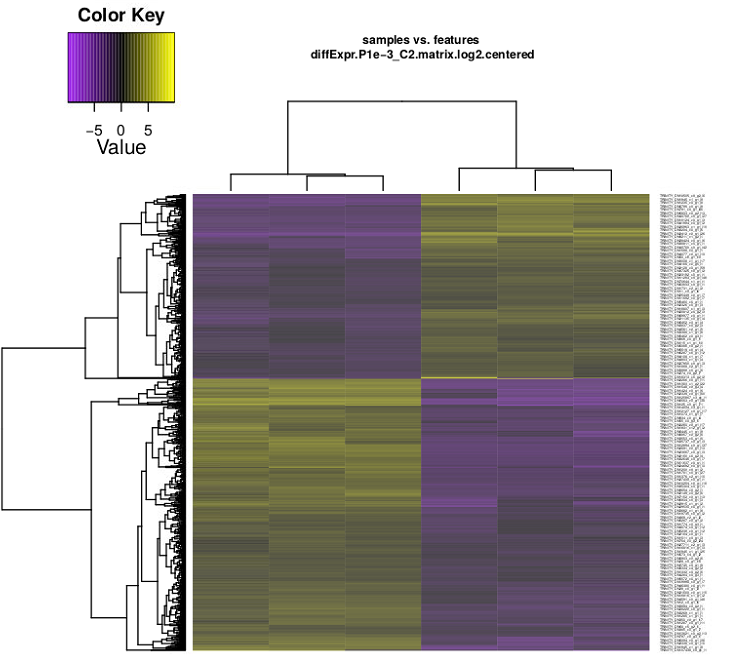

引用
De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis
Brian J Haas, Alexie Papanicolaou, Moran Yassour, Manfred Grabherr, Philip D Blood, Joshua Bowden, Matthew Brian Couger, David Eccles, Bo Li, Matthias Lieber, Matthew D MacManes, Michael Ott, Joshua Orvis, Nathalie Pochet, Francesco Strozzi, Nathan Weeks, Rick Westerman, Thomas William, Colin N Dewey, Robert Henschel, Richard D LeDuc, Nir Friedman , Aviv Regev
Nat Protoc. 2013 Aug;8(8):1494-512