トランスクリプトームのゲノムワイドな解析は、既知のすべての生物種の生理学の基礎となる分子メカニズムに関する広範な洞察を提供し、まだ隠されているものを発見することを可能にする。近年、オックスフォード・ナノポア・テクノロジー(ONT)は、次世代シーケンシングに代わる、高速、小型、ポータブルでコスト効率の高い技術として開発された。しかし、ONTの携帯性を活かし、バイオインフォマティクスの専門知識がなくてもONTのデータをどこでも簡単に解析できるRNA-seqデータ解析ソフトウェアは、広く普及していない。本著者らは、DuesselporeTMを開発した。DuesselporeTMは、ローカルウェブサーバーとして動作し、追加のバイオインフォマティクスツールやインターネット接続を必要とせず、どこでもONTデータの解析を可能にする、わかりやすいディープシーケンサーのワークフローである。DuesselporeTMの出力には、発現変動した遺伝子が含まれており、さらに、分散ヒートマップ、疾患および遺伝子オントロジープロット、遺伝子コンセプトネットワークプロット、異なる細胞プロセスのためのカスタマイズされたパスウェイのエクスポートなどの下流の分析が可能になっている。DuesselporeTMは、ダイオキシン様PCBであり、強力なアリル炭化水素受容体(AhR)アゴニストであるPCB126によって引き起こされるトランスクリプトームの変化を、よく知られたモデルシステムであるヒトHaCaTケラチノサイトで解析することによって検証された。DuesselporeTMは、特にONTデータを解析するために開発されたが、NGSデータの解析も実施した。DuesselporeTMは、MicrosoftおよびMac OSに対応しており、ONTおよびNGSデータの解析を便利に、信頼性高く、コスト効率よく行うことができる。
DuesselporeのWebフレームワークはDjango Python 3.8.5を用いて作成されており、NGSデータ解析機能はRとPythonを用いて作成されている。RNASeqのワークフローは、標準的なデータ処理パイプラインに従っている。まず、FastQCによる品質管理が行われる。配列品質を定量化し、HTMLファイルに出力する。最先端のツールであるMinimap2 (17)を用いて、リードをリファレンスゲノム/トランスクリプトームにマッピングする。アラインされたBAMファイルはRsubread/featureCounts(18)で処理される。HTSeq/htseq-count、またはSalmonで処理し、生のリードカウントを生成する。Salmonはfastqからリファレンスゲノムに直接定量することができるが、Minimap2の方が、ノイズの多いONTデータを扱う際の割り当て率が高い。生のリードカウントマトリクスを、DESeq2, edgeR/limmaを用いて処理する。DESeq2はレプリケーツ数が少ないデータを処理する能力が高い。遺伝子オントロジーの解析には、BioconductorのGene Ontology pipeline、ここではGene Ontologyのパスウェイ(Gene Set Enrichment Analysis of Gene Ontology of Gene Ontology (gseGO), Disease Ontology, Network of Cancer Gene (DOSE), Pathview)が使用されている。
manual.md
duesselpore/manual.md at main · thachnguyen/duesselpore · GitHub
インストール
DuesselporeはあなたのDockerコンテナまたは仮想マシン(Virtualbox)上でローカルに実行できる。ここではdockerを利用した(イメージのpull後はネット接続の必要なし、オフラインのマシンで解析可能)。
ハードウェア
- CPU: 2.0 GHz (Intel architecture, acceleration support) 4 cores or higher (8 cores recommended)
- System memory (RAM): 8 GB or higher (16 GB recommended)
- Diskdrive: 200 GB free space (1TB recommended)
- Host operating system Window 10, Linux (Ubuntu >=18.04 or Fedora) or MacOS (Intel)
依存
- R version 4.1.1
- Bioconductor and its packages Full R packages and session info in R.txt
- Python 3.8.5, required packages in requirements.txt
- minimap2 version 2.20-r1061
- samtools version 1.7
- HTSeq 0.13.5
- Salmon v1.5.2
sudo docker run -it -p 8000:8000 thachdt4/duesselpore:running python3 /home/ag-rossi/projects/duesselpore/manage.py runserver 0.0.0.0:8000
http://localhost:8000/duesselpore にアクセスする。
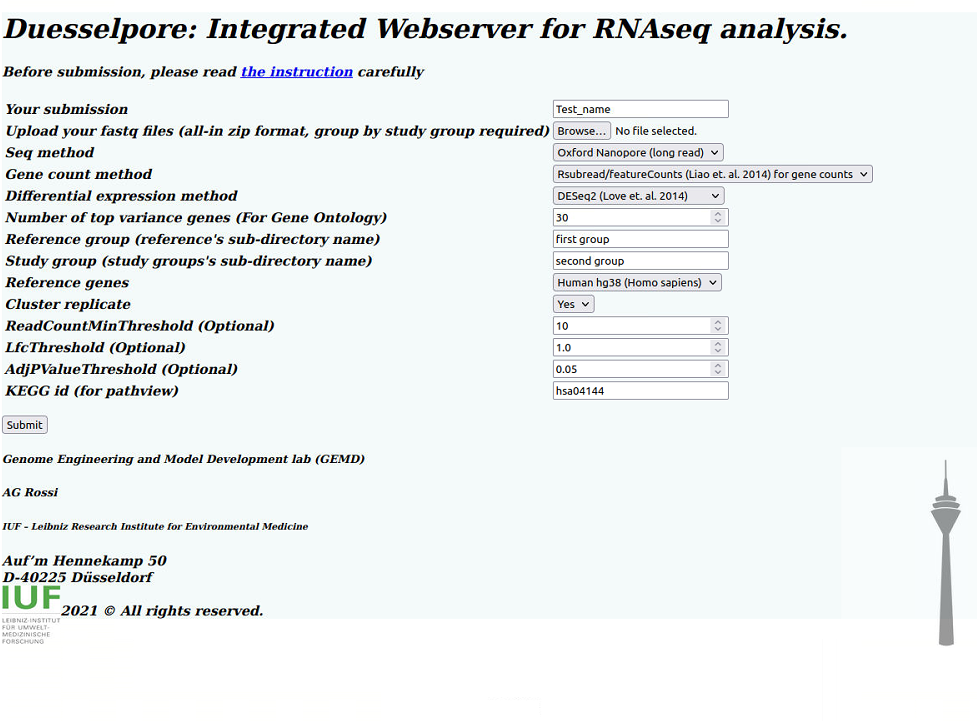
実行方法
マニュアルに書いてあるが、解析するfastqファイルは条件ごとにサブフォルダーを作ってその中に配置する。それから、このディレクトリ(画像ではfastq/)をzipで固めてアップロードする。

(マニュアルより)
zipファイルをアップロードする。ここではマニュアルで公開されている軽量版のテストデータ(軽量バージョンとフルバージョンのリンクがある。軽量バージョンはzip圧縮で2.0GB)を使った。
OXford nanopore long readの他にPacBioのロングリード、illuminaのショートリードが選択できる。

カウント方法。デフォルトはsubreadパッケージ。ここではsalmonに変更した。

他のパラメータ。対照群、実験群のディレクトリ名、リファレンスゲノムなどを確認しておく。


テストデータはhuman。

kegg id名も注意する(デフォルトで記載されているのは、”hsa04144hsa”はヒト(hsa)のEndocytosisパスウェイ(map04144))。複数のpathway IDは記載できない。

submitボタンを押しても画面に変化はないが、すぐにzipの解凍が始まり、続いて計算がスタートする。軽量テストデータの場合は1.5時間かかるとされる(著者らの計算環境)。
出力例
計算が完了すると、すべての結果がブラウザからダウンロードできるようになる(画像右上のDownload results)。ただし、図はダウンロード前の方がインタラクティブに操作可能。
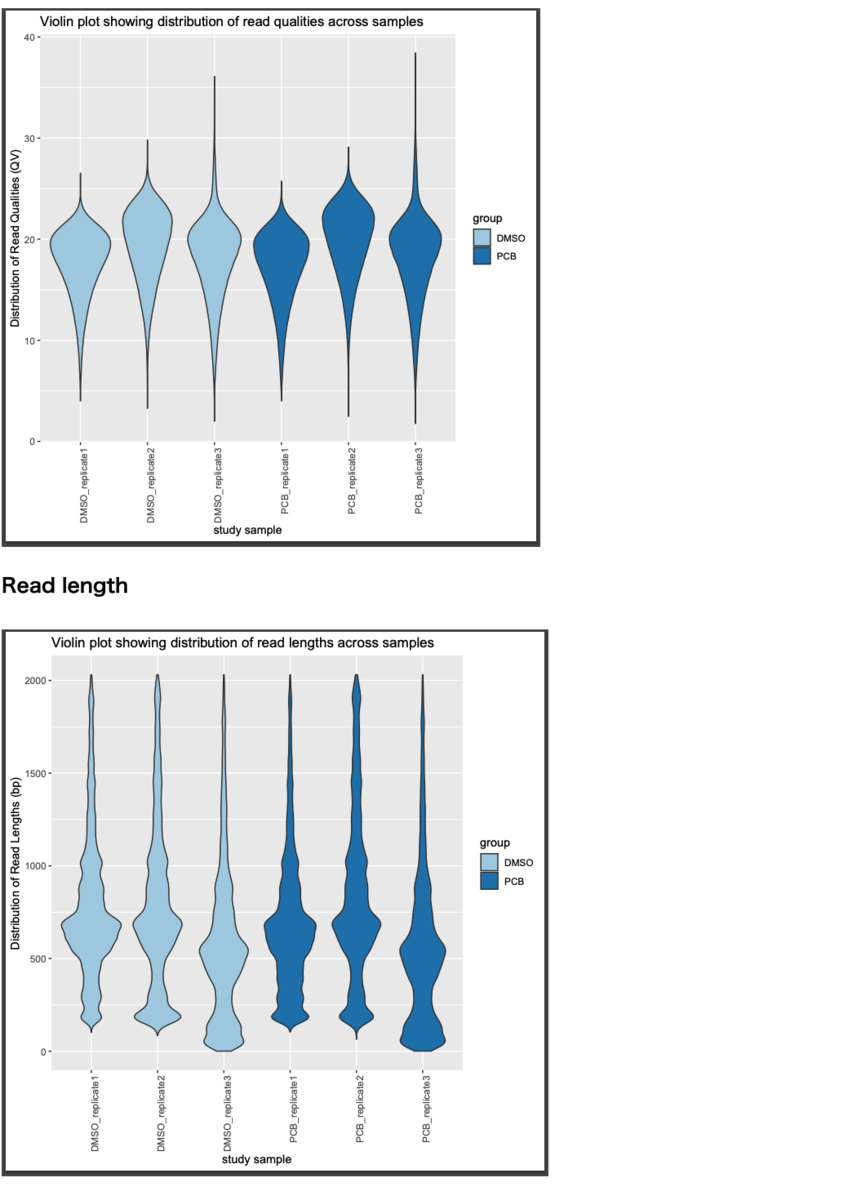


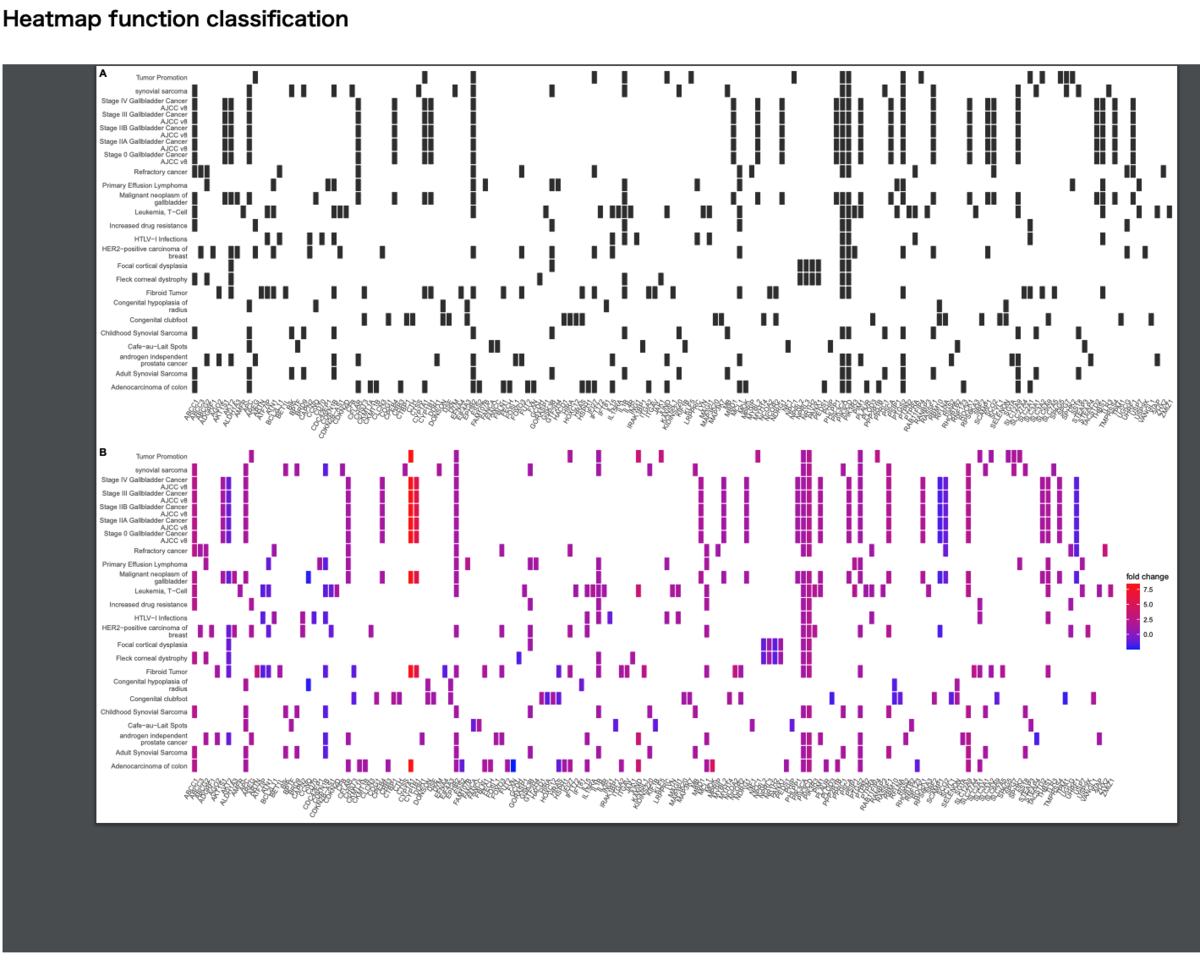
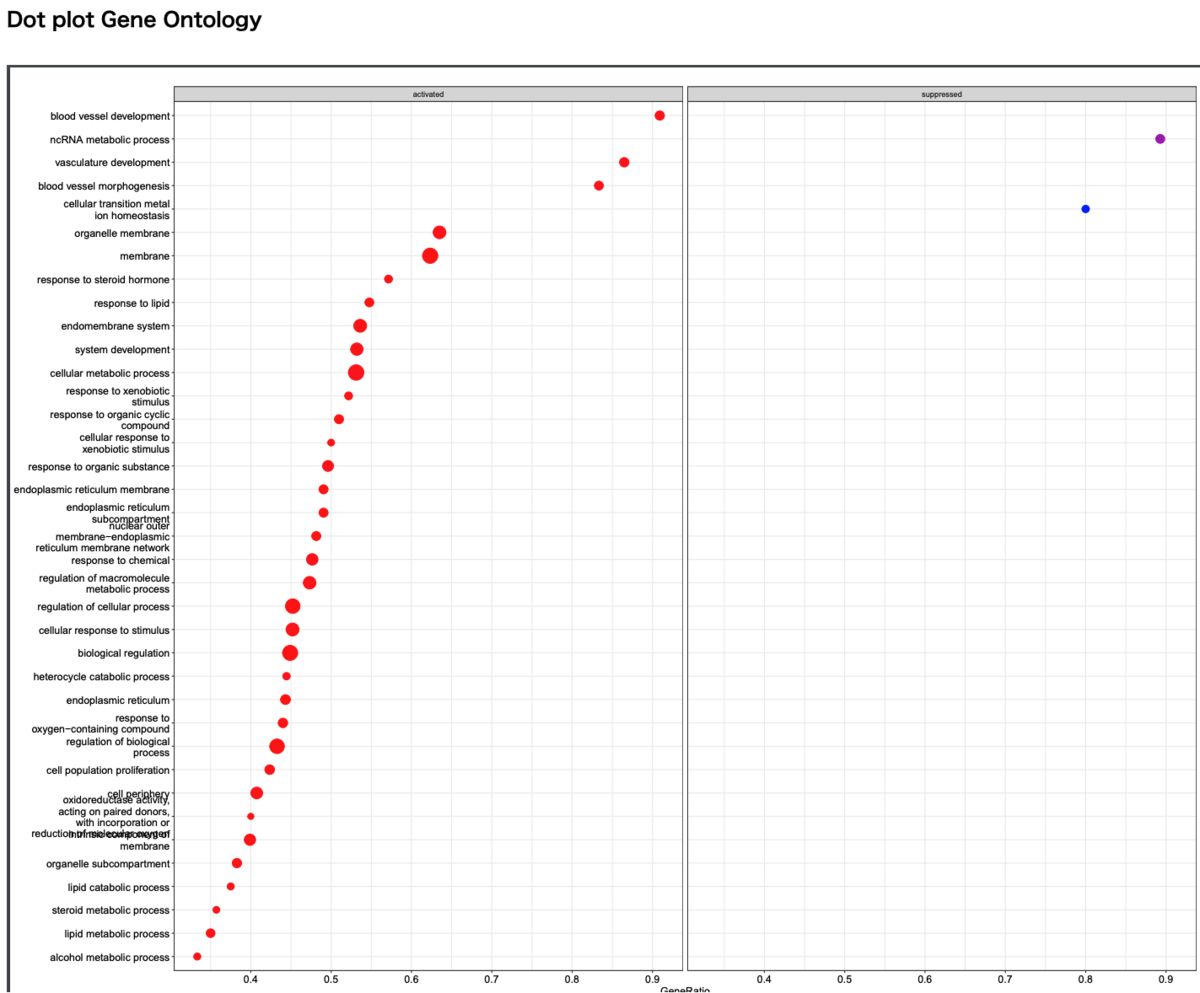
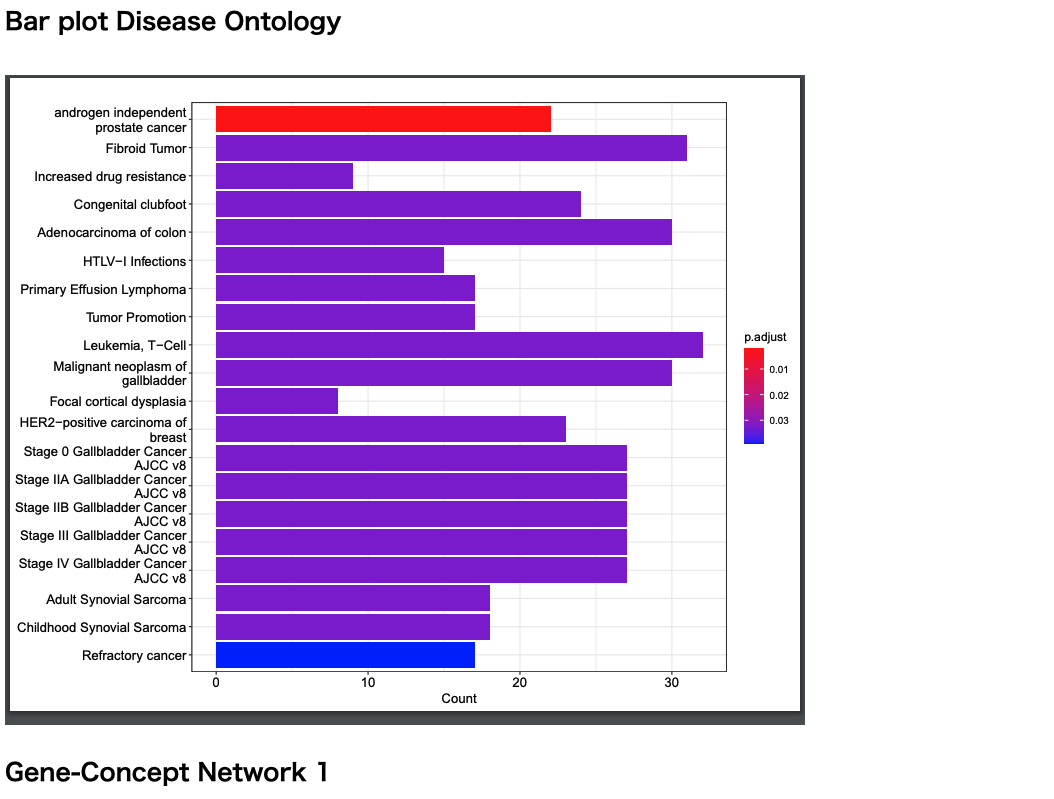
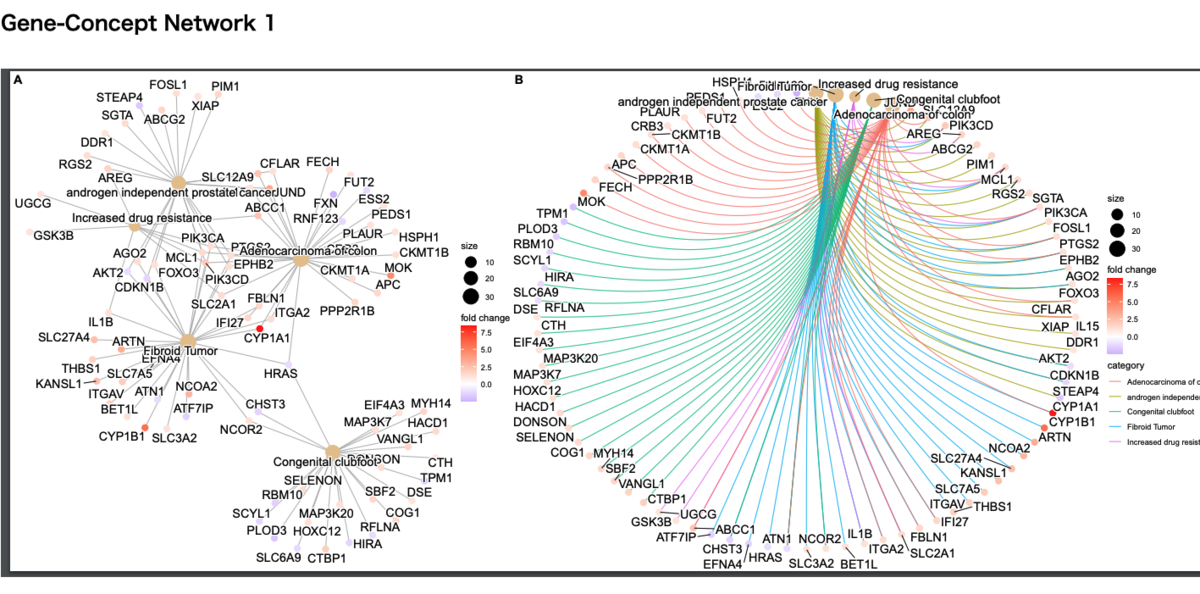
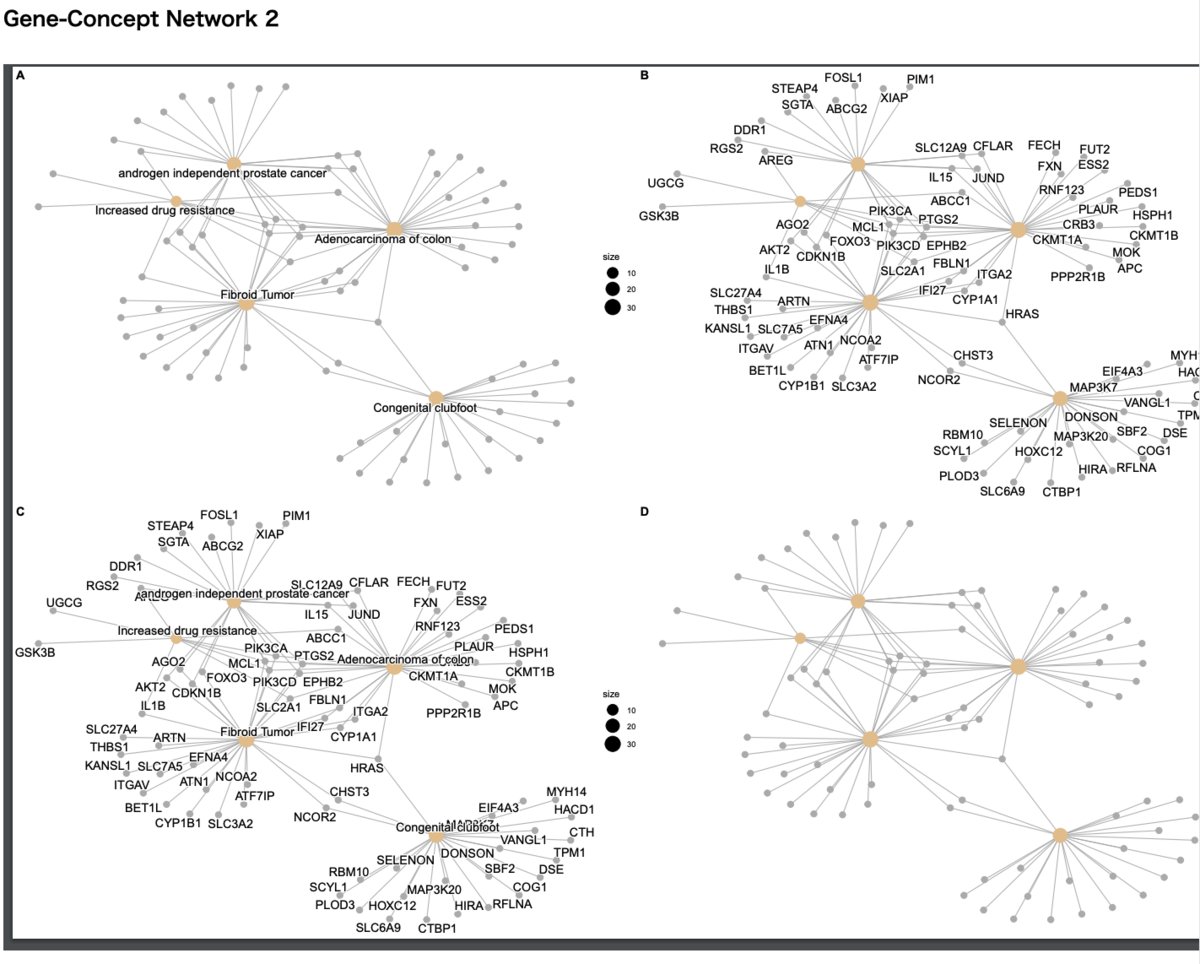
軽量テストデータでは、一部の解析が正常に終了しなかった。
ダウンロードしたファイル(軽量データ)

引用
Duesselpore: a full-stack local web server for rapid and simple analysis of Oxford Nanopore Sequencing data
Christian Vogeley, Thach Nguyen, Selina Woeste, Jean Krutmann, Thomas Haarmann-Stemmann, Andrea Rossi
bioRxiv, Posted November 16, 2021