2020 11/30 文章修正、help追加、condaによるインストールコマンド追加
TransRateは、de novoトランスクリプトームアセンブリの品質をリファレンスフリーで評価するためのツールである。シークエンシングリードとアセンブリのみを入力として使用して、de novoトランスクリプトームアセンブリの複数の一般的なアーチファクトを容易に検出できる。これらには、キメラ、構造エラー、不完全なアセンブリ、塩基エラーなどが含まれる。TransRateはこれらのエラーを評価して各コンティグの診断品質スコアを作成し、これらのコンティグスコアを統合してアセンブリ全体を評価する。このように、TransRateは、de novoアセンブリのフィルタリングや最適化だけでなく、同じ入力リードから異なる方法で生成されたアセンブリの比較にも使用することができる。公開されている155のde novoトランスクリプトームアセンブリデータセットにこの手法を適用し、アセンブリの方法、リード長、リード量、リード品質がde novoトランスクリプトームアセンブリの精度に与える寄与を分解したところ、入力データの品質のばらつきが公開されているde novoトランスクリプトームアセンブリーの品質のばらつきの43%を説明していることが明らかになった。TransRateはリファレンスフリーなので、ロングノンコーディングRNA、rRNA、mRNA、混合RNAサンプルのアセンブリーを含む、あらゆるタイプのRNAのアセンブリーの評価に適している。
Translateはde novo transcriptomeの精度をリードのアライメントのされ方などから評価するツール。発表は2016年だが、すでにいくつかのペーパーに引用されている。BUSCOとTransRateでcore gene数とエラー率を見積もり、アセンブルの精度を担保した上で進めることが今後のde novo transcriptome解析の常套手段になるかもしれない。導入して動作をテストしていく。
マニュアル
Transrate - transcriptome assembly quality analysis
インストール
依存
- blast+
- snap-aligner #リードのアライメントに使う
- bam-read
- Salmon
#bioconda (link)
conda install -c bioconda -y transrate
#他の依存
transrate --install-deps all
#dockerhub(link)
docker pull arnaudmeng/transrate:1.0.3
---condaを使わない場合---
brewでbam-read以外は導入できる。
brew install snap-aligner
brew install blast
brew install salmon
bam-readのダウンロード直リンク (TransRateマニュアルより)
解凍すると本体のbinaryのみできる。パスが通っている場所に移動させる。
chmod u+x #実行権がなければ
sudo cp -i bam-read /usr/locan/bin/ #binに移動
本体
Transrate - transcriptome assembly quality analysis
Binaryのみダウンロードできる。ファイル構造を壊すと動かなくなるので注意する。パスを通しておく。
echo "export PATH=$PATH:/Users/kazumaxneo/Documents/transrate-1.0.3-osx/" >> ~/.bash_profile #環境に合わせて変えてください
source ~/.bash_profile #ソースする
> transrate
Transrate v1.0.3
by Richard Smith-Unna, Chris Boursnell, Rob Patro,
Julian Hibberd, and Steve Kelly
DESCRIPTION:
Analyse a de-novo transcriptome assembly using three kinds of metrics:
1. sequence based (if --assembly is given)
2. read mapping based (if --left and --right are given)
3. reference based (if --reference is given)
Documentation at http://hibberdlab.com/transrate
USAGE:
transrate <options>
OPTIONS:
--assembly=<s> Assembly file(s) in FASTA format, comma-separated
--left=<s> Left reads file(s) in FASTQ format, comma-separated
--right=<s> Right reads file(s) in FASTQ format, comma-separated
--reference=<s> Reference proteome or transcriptome file in FASTA format
--threads=<i> Number of threads to use (default: 8)
--merge-assemblies=<s> Merge best contigs from multiple assemblies into file
--output=<s> Directory where results are output (will be created) (default: transrate_results)
--loglevel=<s> Log level. One of [error, info, warn, debug] (default: info)
--install-deps=<s> Install any missing dependencies. One of [read, ref, all]
--examples Show some example commands with explanations
docker
docker run --rm -itv $PWD:/data -w /data arnaudmeng/transrate:1.0.3
transrate -h
準備
Transrate - transcriptome assembly quality analysisから、テストファイルをダウンロードして解凍する。
3つファイルができる。

ラン
1、contigの基本統計を得る
de novo assemblyして得たtranscripts.fastaを分析する。
transrate --assembly transcripts.fa --threads 12
transrate_results/にCSVファイルのassemblies.csvとtrasncripts/contigs.csvができる。
assemblies.csvには配列の長さなどの基本情報が含まれている。Nの数や、GC含量のほか、アセンブリの指標に用いられるN50、N70なども計算されている。
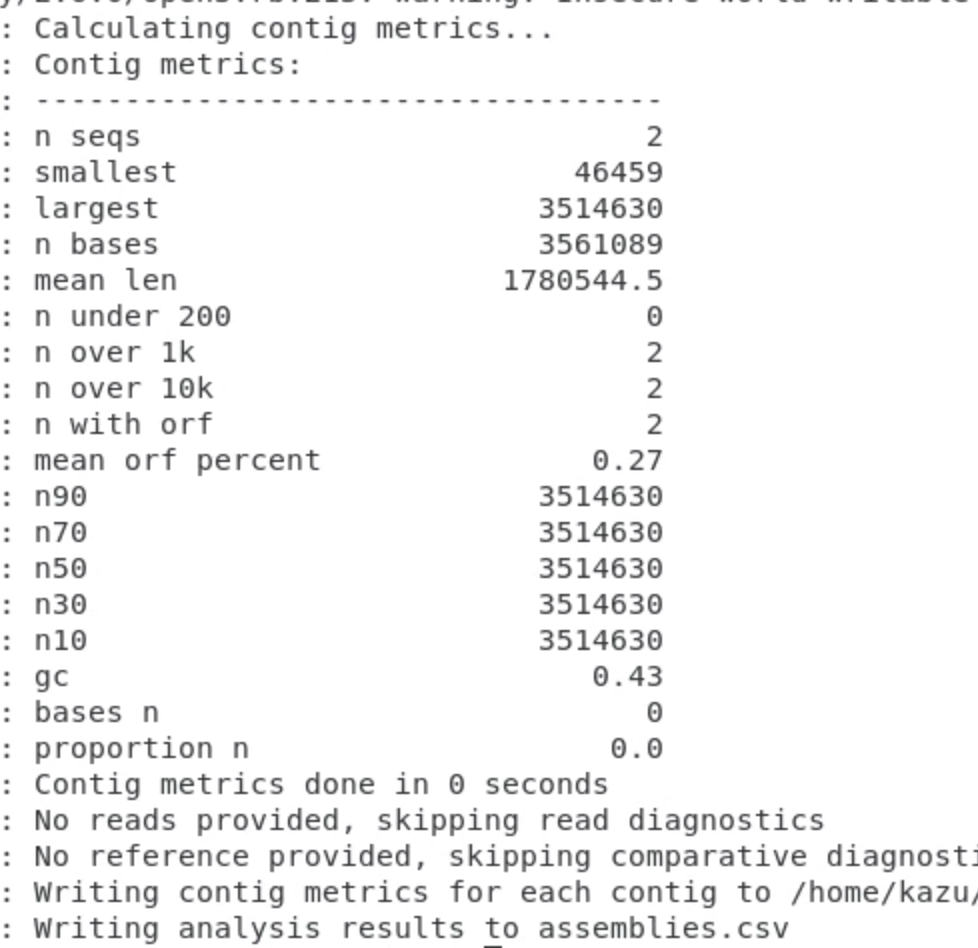
詳細はTransrate - transcriptome assembly quality analysisのContig metricsを参照。
contigs.csvはcontigそれぞれの長さ、GC含量が計算、出力されている。columnコマンドに-sをつけて仕切りをCSVフォーマットの","と明示している。
user$ column -t -s',' transcripts/contigs.csv
contig_name length prop_gc orf_length
NM_001168316 2283 0.487516 533
NM_174914 2385 0.485535 567
NR_031764 1853 0.507285 221
NM_004503 1681 0.527662 235
NM_006897 1541 0.552239 260
NM_014212 2037 0.566519 304
NM_014620 2300 0.508261 264
NM_017409 1959 0.533435 342
NM_017410 2396 0.57596 330
NM_018953 1612 0.591811 229
NM_022658 2288 0.482517 242
NM_153633 1666 0.493998 279
NM_153693 2072 0.527992 153
NM_173860 849 0.666667 282
NR_003084 1640 0.560366 182
このようにアセンブルして作ったcontigの要約統計を得ることができる。
2、リード使いアセンブル精度を推測する。
シーケンスリードを入力する。
transrate --assembly transcripts.fa --left left.fq --right right.fq --threads 20
- --threads thread number
リード数が多いと少し時間がかかる(e.g., 10GBx2のデータでCPU30指定だと1時間弱)。
やっていることについては公式ページのRead mapping metricsを参考。解析が終了すると、標準出力にまとめの表が表示される。

例えば"n bases uncovered"はリードのマッピングがゼロになっている塩基の数である。詳細は公式マニュアルのRead mapping metrics 参照。
ここで上の画面の下の方にあるTransrate assembly scoreに注目する。
TRANSRATE ASSEMBLY SCORE 0.6701
リードを指定した時だけこのTransrate assembly scoreは計算される。Transrate assembly scoreはアセンブルデータ全体と各contigについて計算されていて、ここでは全体のスコアを見ている。高い方がベターで、1が最高である。よくこの数値がアセンブルの指標として論文で比較されたりする(比較が可能なのは、同一のシーケンスリードで評価したアセンブルデータ)。
他にcontig score、optimised assembly scoreなどがなる。詳細は論文と公式ページ(リンク)で確認してください。
出力ディレクトリには他にもたくさんのファイルができる。good.transcripts.faとbad..transcripts.faは、ペアリードの両方がアライメントされ、且つ向きも正しいか(--> <--)、contig末端がオーバーラップしていないかなどを指標にcontig-scoreが出され、そのスコアにより選別されている。test dataでは10000 contingのうち6 がbadであった。
補足
シーケンスリードによるアセンブルデータの分析は、使うシーケンスリードの精度に100%依存している。例えば使用したリードがlow qualityだったりアダプター配列が付いたままだったりすると、結果は大きく影響を受けてしまうので、オーサーらはアダプター配列トリミングとクオリティトリミングを行い、かつエラーコレクションも終わったデータを使うことを勧めている。
エラーコレクションは、例えばrnaspadesならBayesHammerでエラーコレクションされたリードがrnaspades出力ディレクトリのcorrect/にgz形式で保存されている。このようなリードを使うことを推薦している(注; エラーコレクションはRNA用のものを使う必要がある。ゲノム用のエラーコレクションを使うと、k-mer coverageが低い低発現の情報が軒並み除去される恐れがある)。
リード数を(diginormなどで)ノーマライズしたデータを使う点については、公式サイトは使っても影響はないと主張している。ただしここではそういったことは行わない。 (蛇足)
transcripts.fasta_bam_info.csvの中身

goodかbadの指標になるデータがまとめられている。
contigs.csvの中身

先ほど標準出力された内容がcontigごとにまとめられている。
複数のfastaを同時に調べる。
transrate --assembly one.fa,two.fa
”," でサンプルを繋ぐ。
4、モデル生物の情報からアセンブル精度を推測する。
transrate --assembly transcripts.fa --reference ref.fa
de novo transcriptomeならリファレンスのcDNA情報はないことになるが、近縁種(very closely)のゲノム情報が利用できるなら、それと比較してアセンブリ精度を確認することもできる。比較はblastで相同性を調べることで行われる。
補足
ここでref.faはシロイヌナズナのcDNA配列を指定しているが、植物では1種類の遺伝子に多くのパラログがあったりして、複数ヒットによりスコアが不当に低くなってしまうことが起こる。公式サイトは、single のtranscriptに絞った配列をJGIのphytozomeなどから入手し、それをリファンレスにすることを勧めている(Choosing a reference)。
解析が終了すると、標準出力にまとめの表が表示される。

詳細は公式マニュアルのComparative metrics 参照。
transcripts_into_Arabidopsis_thaliana.TAIR10.cdna.all.1.blast

各々のcontigのblast結果がまとめられている。
contigs.csvの中身

先ほど標準出力された内容がcontigごとにまとめられている。
下のようなエラーが出た場合、
/home/transrate-1.0.3-linux-x86_64/lib/app/ruby/lib/ruby/gems/2.2.0/gems/bundler-1.7.12/lib/bundler/runtime.rb:222: warning: Insecure world writable dir /usr/local/bin/contig_Hunter/ in PATH, mode 040777
Dependencies are missing:
- blastplus (2.2.[0-9])
transrate --install-deps ref
で依存パッケージをインストール(公式サイト説明 一番下)。
5、リードとリファンレンスを両方使いアセンブル精度を推測する。
transrate --assembly transcripts.fa --left left.fq --right right.fq --threads 12 --reference ref.fa
どのデータ使うかは、その解析によって変わってくる。最近nature Scientific Reports に発表された論文(リンク Table. 2)では、以下の8つの情報を使っている。
シーケンスリードから計算されるRead mapping metrics (#2)
- % fragments mapped
- % good mappings
- % bases uncovered
リファンレンスから計算されるComparative metrics (#3)
- % contigs with CRBB
- % refs with CRBB
- Reference coverage
総合的なスコア; TransRate score (#2)
- TransRate assembly score
- % good contigs
コマンドだけ打つとヘルプが出る。
transrate
_ _
| |_ _ __ __ _ _ __ ___ _ __ __ _ | |_ ___
░▓▓▓^▓▓▓░ | __|| '__|/ _` || '_ \ / __|| '__|/ _` || __|/ _ \ ░▓▓▓^▓▓▓░
░▓▓▓^▓▓▓░ | |_ | | | (_| || | | |\__ \| | | (_| || |_| __/ ░▓▓▓^▓▓▓░
░▓▓▓^▓▓▓░ \__||_| \__,_||_| |_||___/|_| \__,_| \__|\___| ░▓▓▓^▓▓▓░
Transrate v1.0.3
by Richard Smith-Unna, Chris Boursnell, Rob Patro,
Julian Hibberd, and Steve Kelly
DESCRIPTION:
Analyse a de-novo transcriptome assembly using three kinds of metrics:
1. sequence based (if --assembly is given)
2. read mapping based (if --left and --right are given)
3. reference based (if --reference is given)
Documentation at http://hibberdlab.com/transrate
USAGE:
transrate <options>
OPTIONS:
--assembly=<s> Assembly file(s) in FASTA format, comma-separated
--left=<s> Left reads file(s) in FASTQ format, comma-separated
--right=<s> Right reads file(s) in FASTQ format, comma-separated
--reference=<s> Reference proteome or transcriptome file in FASTA format
--threads=<i> Number of threads to use (default: 8)
--merge-assemblies=<s> Merge best contigs from multiple assemblies into file
--output=<s> Directory where results are output (will be created) (default: transrate_results)
--loglevel=<s> Log level. One of [error, info, warn, debug] (default: info)
--install-deps=<s> Install any missing dependencies. One of [ref]
--examples Show some example commands with explanations
uesaka-no-MacBook-Air-2:transrate-1.0.3-osx kazumaxneo$
ユーザー目線で作られている。素晴らしい。
#注 リード数が多すぎると(150GBx2)計算が終わらない事がありました。その時はサブサンプリングしてリード数を減らすとランできました。
- 配列数が数十万を超えるとsnapでエラーを起こすことがある。goodとbadの配列に分離したい場合、サブセットに対してコマンドを繰り返すか、先にほかのフィルタリングを実行して配列数を減らす。
引用
TransRate: reference-free quality assessment of de novo transcriptome assemblies.
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M. & Kelly, S.
Genome Res. 26, 1134–1144 (2016).
TransRateとBUSCOを使ってアセンブリを評価している論文
https://www.nature.com/articles/sdata2016131.pdf