2019 4/16 condaインストール
2019 12/9ビルド手順の誤り修正
2020 1/5 mergeの説明追加
2020 4/18 基本コマンド追記
2020 10/13 追記
20200 10/14 分かりにくい説明を修正
2021 2/17 dockerリンク追加
2021 5/16 ”変異”を”バリアント”に修正
2023/09/29 vcffilterの例を修正
VCFtoolsは、バリアントコールフォーマットのVCFファイルのマージ、ソートやフィルタリング、固有変異の抽出などができるツール。 よく使いそうなコマンドに限って紹介する。
HP
マニュアル
https://vcftools.github.io/perl_module.html
インストール
#linux
git clone https://github.com/vcftools/vcftools.git
cd vcftools
./autogen.sh
./configure --prefix=/usr/local/
make -j
make install
#Bioconda (link)mac, linux。コマンドが違うので注意。上の手順の通り、gitからcloneしてビルドして下さい。
conda install -c bioconda -y vcftools
#2021 2/17 dockerhub (link)
docker pull biocontainers/vcftools:v0.1.16-1-deb_cv1
マニュアル
man vcftools
vcftools(man) 05 January 2016 vcftools(man)
NAME
vcftools v0.1.14 - Utilities for the variant call format (VCF) and binary variant call format (BCF)
SYNOPSIS
vcftools [ --vcf FILE | --gzvcf FILE | --bcf FILE] [ --out OUTPUT PREFIX ] [ FILTERING OPTIONS ] [ OUTPUT OPTIONS ]
DESCRIPTION
vcftools is a suite of functions for use on genetic variation data in the form of VCF and BCF files. The tools provided will be used mainly to summarize data, run calculations on data, filter out data, and convert data into other useful file formats.
EXAMPLES
Output allele frequency for all sites in the input vcf file from chromosome 1
vcftools --gzvcf input_file.vcf.gz --freq --chr 1 --out chr1_analysis
Output a new vcf file from the input vcf file that removes any indel sites
vcftools --vcf input_file.vcf --remove-indels --recode --recode-INFO-all --out SNPs_only
Output file comparing the sites in two vcf files
vcftools --gzvcf input_file1.vcf.gz --gzdiff input_file2.vcf.gz --diff-site --out in1_v_in2
Output a new vcf file to standard out without any sites that have a filter tag, then compress it with gzip
vcftools --gzvcf input_file.vcf.gz --remove-filtered-all --recode --stdout | gzip -c > output_PASS_only.vcf.gz
Output a Hardy-Weinberg p-value for every site in the bcf file that does not have any missing genotypes
vcftools --bcf input_file.bcf --hardy --max-missing 1.0 --out output_noMissing
Output nucleotide diversity at a list of positions
zcat input_file.vcf.gz | vcftools --vcf - --site-pi --positions SNP_list.txt --out nucleotide_diversity
BASIC OPTIONS
These options are used to specify the input and output files.
INPUT FILE OPTIONS
--vcf <input_filename>
This option defines the VCF file to be processed. VCFtools expects files in VCF format v4.0, v4.1 or v4.2. The latter two are supported with some small limitations. If the user provides a dash character '-' as a file name, the program expects a VCF file to
be piped in through standard in.
--gzvcf <input_filename>
This option can be used in place of the --vcf option to read compressed (gzipped) VCF files directly.
--bcf <input_filename>
This option can be used in place of the --vcf option to read BCF2 files directly. You do not need to specify if this file is compressed with BGZF encoding. If the user provides a dash character '-' as a file name, the program expects a BCF2 file to be piped
in through standard in.
OUTPUT FILE OPTIONS
--out <output_prefix>
This option defines the output filename prefix for all files generated by vcftools. For example, if <prefix> is set to output_filename, then all output files will be of the form output_filename.*** . If this option is omitted, all output files will have the
prefix "out." in the current working directory.
--stdout
-c
These options direct the vcftools output to standard out so it can be piped into another program or written directly to a filename of choice. However, a select few output functions cannot be written to standard out.
--temp <temporary_directory>
This option can be used to redirect any temporary files that vcftools creates into a specified directory.
SITE FILTERING OPTIONS
These options are used to include or exclude certain sites from any analysis being performed by the program.
POSITION FILTERING
--chr <chromosome>
--not-chr <chromosome>
Includes or excludes sites with indentifiers matching <chromosome>. These options may be used multiple times to include or exclude more than one chromosome.
--from-bp <integer>
--to-bp <integer>
These options specify a lower bound and upper bound for a range of sites to be processed. Sites with positions less than or greater than these values will be excluded. These options can only be used in conjunction with a single usage of --chr. Using one of
these does not require use of the other.
--positions <filename>
--exclude-positions <filename>
Include or exclude a set of sites on the basis of a list of positions in a file. Each line of the input file should contain a (tab-separated) chromosome and position. The file can have comment lines that start with a "#", they will be ignored.
--positions-overlap <filename>
--exclude-positions-overlap <filename>
Include or exclude a set of sites on the basis of the reference allele overlapping with a list of positions in a file. Each line of the input file should contain a (tab-separated) chromosome and position. The file can have comment lines that start with a
"#", they will be ignored.
--bed <filename>
--exclude-bed <filename>
Include or exclude a set of sites on the basis of a BED file. Only the first three columns (chrom, chromStart and chromEnd) are required. The BED file is expected to have a header line. A site will be kept or excluded if any part of any allele (REF or ALT)
at a site is within the range of one of the BED entries.
--thin <integer>
Thin sites so that no two sites are within the specified distance from one another.
--mask <filename>
--invert-mask <filename>
--mask-min <integer>
These options are used to specify a FASTA-like mask file to filter with. The mask file contains a sequence of integer digits (between 0 and 9) for each position on a chromosome that specify if a site at that position should be filtered or not.
An example mask file would look like:
>1
0000011111222...
>2
2222211111000...
In this example, sites in the VCF file located within the first 5 bases of the start of chromosome 1 would be kept, whereas sites at position 6 onwards would be filtered out. And sites after the 11th position on chromosome 2 would be filtered out as well.
The "--invert-mask" option takes the same format mask file as the "--mask" option, however it inverts the mask file before filtering with it.
And the "--mask-min" option specifies a threshold mask value between 0 and 9 to filter positions by. The default threshold is 0, meaning only sites with that value or lower will be kept.
SITE ID FILTERING
--snp <string>
Include SNP(s) with matching ID (e.g. a dbSNP rsID). This command can be used multiple times in order to include more than one SNP.
--snps <filename>
--exclude <filename>
Include or exclude a list of SNPs given in a file. The file should contain a list of SNP IDs (e.g. dbSNP rsIDs), with one ID per line. No header line is expected.
VARIANT TYPE FILTERING
--keep-only-indels
--remove-indels
Include or exclude sites that contain an indel. For these options "indel" means any variant that alters the length of the REF allele.
FILTER FLAG FILTERING
--remove-filtered-all
Removes all sites with a FILTER flag other than PASS.
--keep-filtered <string>
--remove-filtered <string>
Includes or excludes all sites marked with a specific FILTER flag. These options may be used more than once to specify multiple FILTER flags.
INFO FIELD FILTERING
--keep-INFO <string>
--remove-INFO <string>
Includes or excludes all sites with a specific INFO flag. These options only filter on the presence of the flag and not its value. These options can be used multiple times to specify multiple INFO flags.
ALLELE FILTERING
--maf <float>
--max-maf <float>
Include only sites with a Minor Allele Frequency greater than or equal to the "--maf" value and less than or equal to the "--max-maf" value. One of these options may be used without the other. Allele frequency is defined as the number of times an allele
appears over all individuals at that site, divided by the total number of non-missing alleles at that site.
--non-ref-af <float>
--max-non-ref-af <float>
--non-ref-ac <integer>
--max-non-ref-ac <integer>
--non-ref-af-any <float>
--max-non-ref-af-any <float>
--non-ref-ac-any <integer>
--max-non-ref-ac-any <integer>
Include only sites with all Non-Reference (ALT) Allele Frequencies (af) or Counts (ac) within the range specified, and including the specified value. The default options require all alleles to meet the specified criteria, whereas the options appended with
"any" require only one allele to meet the criteria. The Allele frequency is defined as the number of times an allele appears over all individuals at that site, divided by the total number of non-missing alleles at that site.
--mac <integer>
--max-mac <integer>
Include only sites with Minor Allele Count greater than or equal to the "--mac" value and less than or equal to the "--max-mac" value. One of these options may be used without the other. Allele count is simply the number of times that allele appears over
all individuals at that site.
--min-alleles <integer>
--max-alleles <integer>
Include only sites with a number of alleles greater than or equal to the "--min-alleles" value and less than or equal to the "--max-alleles" value. One of these options may be used without the other.
For example, to include only bi-allelic sites, one could use:
vcftools --vcf file1.vcf --min-alleles 2 --max-alleles 2
GENOTYPE VALUE FILTERING
--min-meanDP <float>
--max-meanDP <float>
Includes only sites with mean depth values (over all included individuals) greater than or equal to the "--min-meanDP" value and less than or equal to the "--max-meanDP" value. One of these options may be used without the other. These options require that
the "DP" FORMAT tag is included for each site.
--hwe <float>
Assesses sites for Hardy-Weinberg Equilibrium using an exact test, as defined by Wigginton, Cutler and Abecasis (2005). Sites with a p-value below the threshold defined by this option are taken to be out of HWE, and therefore excluded.
--max-missing <float>
Exclude sites on the basis of the proportion of missing data (defined to be between 0 and 1, where 0 allows sites that are completely missing and 1 indicates no missing data allowed).
--max-missing-count <integer>
Exclude sites with more than this number of missing genotypes over all individuals.
--phased
Excludes all sites that contain unphased genotypes.
MISCELLANEOUS FILTERING
--minQ <float>
Includes only sites with Quality value above this threshold.
INDIVIDUAL FILTERING OPTIONS
These options are used to include or exclude certain individuals from any analysis being performed by the program.
--indv <string>
--remove-indv <string>
Specify an individual to be kept or removed from the analysis. This option can be used multiple times to specify multiple individuals. If both options are specified, then the "--indv" option is executed before the "--remove-indv option".
--keep <filename>
--remove <filename>
Provide files containing a list of individuals to either include or exclude in subsequent analysis. Each individual ID (as defined in the VCF headerline) should be included on a separate line. If both options are used, then the "--keep" option is executed
before the "--remove" option. When multiple files are provided, the union of individuals from all keep files subtracted by the union of individuals from all remove files are kept. No header line is expected.
--max-indv <integer>
Randomly thins individuals so that only the specified number are retained.
GENOTYPE FILTERING OPTIONS
These options are used to exclude genotypes from any analysis being performed by the program. If excluded, these values will be treated as missing.
--remove-filtered-geno-all
Excludes all genotypes with a FILTER flag not equal to "." (a missing value) or PASS.
--remove-filtered-geno <string>
Excludes genotypes with a specific FILTER flag.
--minGQ <float>
Exclude all genotypes with a quality below the threshold specified. This option requires that the "GQ" FORMAT tag is specified for all sites.
--minDP <float>
--maxDP <float>
Includes only genotypes greater than or equal to the "--minDP" value and less than or equal to the "--maxDP" value. This option requires that the "DP" FORMAT tag is specified for all sites.
OUTPUT OPTIONS
These options specify which analyses or conversions to perform on the data that passed through all specified filters.
OUTPUT ALLELE STATISTICS
--freq
--freq2
Outputs the allele frequency for each site in a file with the suffix ".frq". The second option is used to suppress output of any information about the alleles.
--counts
--counts2
Outputs the raw allele counts for each site in a file with the suffix ".frq.count". The second option is used to suppress output of any information about the alleles.
--derived
For use with the previous four frequency and count options only. Re-orders the output file columns so that the ancestral allele appears first. This option relies on the ancestral allele being specified in the VCF file using the AA tag in the INFO field.
OUTPUT DEPTH STATISTICS
--depth
Generates a file containing the mean depth per individual. This file has the suffix ".idepth".
--site-depth
Generates a file containing the depth per site summed across all individuals. This output file has the suffix ".ldepth".
--site-mean-depth
Generates a file containing the mean depth per site averaged across all individuals. This output file has the suffix ".ldepth.mean".
--geno-depth
Generates a (possibly very large) file containing the depth for each genotype in the VCF file. Missing entries are given the value -1. The file has the suffix ".gdepth".
OUTPUT LD STATISTICS
--hap-r2
Outputs a file reporting the r2, D, and D' statistics using phased haplotypes. These are the traditional measures of LD often reported in the population genetics literature. The output file has the suffix ".hap.ld". This option assumes that the VCF input
file has phased haplotypes.
--geno-r2
Calculates the squared correlation coefficient between genotypes encoded as 0, 1 and 2 to represent the number of non-reference alleles in each individual. This is the same as the LD measure reported by PLINK. The D and D' statistics are only available for
phased genotypes. The output file has the suffix ".geno.ld".
--geno-chisq
If your data contains sites with more than two alleles, then this option can be used to test for genotype independence via the chi-squared statistic. The output file has the suffix ".geno.chisq".
--hap-r2-positions <positions list file>
--geno-r2-positions <positions list file>
Outputs a file reporting the r2 statistics of the sites contained in the provided file verses all other sites. The output files have the suffix ".list.hap.ld" or ".list.geno.ld", depending on which option is used.
--ld-window <integer>
This optional parameter defines the maximum number of SNPs between the SNPs being tested for LD in the "--hap-r2", "--geno-r2", and "--geno-chisq" functions.
--ld-window-bp <integer>
This optional parameter defines the maximum number of physical bases between the SNPs being tested for LD in the "--hap-r2", "--geno-r2", and "--geno-chisq" functions.
--ld-window-min <integer>
This optional parameter defines the minimum number of SNPs between the SNPs being tested for LD in the "--hap-r2", "--geno-r2", and "--geno-chisq" functions.
--ld-window-bp-min <integer>
This optional parameter defines the minimum number of physical bases between the SNPs being tested for LD in the "--hap-r2", "--geno-r2", and "--geno-chisq" functions.
--min-r2 <float>
This optional parameter sets a minimum value for r2, below which the LD statistic is not reported by the "--hap-r2", "--geno-r2", and "--geno-chisq" functions.
--interchrom-hap-r2
--interchrom-geno-r2
Outputs a file reporting the r2 statistics for sites on different chromosomes. The output files have the suffix ".interchrom.hap.ld" or ".interchrom.geno.ld", depending on the option used.
OUTPUT TRANSITION/TRANSVERSION STATISTICS
--TsTv <integer>
Calculates the Transition / Transversion ratio in bins of size defined by this option. Only uses bi-allelic SNPs. The resulting output file has the suffix ".TsTv".
--TsTv-summary
Calculates a simple summary of all Transitions and Transversions. The output file has the suffix ".TsTv.summary".
--TsTv-by-count
Calculates the Transition / Transversion ratio as a function of alternative allele count. Only uses bi-allelic SNPs. The resulting output file has the suffix ".TsTv.count".
--TsTv-by-qual
Calculates the Transition / Transversion ratio as a function of SNP quality threshold. Only uses bi-allelic SNPs. The resulting output file has the suffix ".TsTv.qual".
--FILTER-summary
Generates a summary of the number of SNPs and Ts/Tv ratio for each FILTER category. The output file has the suffix ".FILTER.summary".
OUTPUT NUCLEOTIDE DIVERGENCE STATISTICS
--site-pi
Measures nucleotide divergency on a per-site basis. The output file has the suffix ".sites.pi".
--window-pi <integer>
--window-pi-step <integer>
Measures the nucleotide diversity in windows, with the number provided as the window size. The output file has the suffix ".windowed.pi". The latter is an optional argument used to specify the step size in between windows.
OUTPUT FST STATISTICS
--weir-fst-pop <filename>
This option is used to calculate an Fst estimate from Weir and Cockerham's 1984 paper. This is the preferred calculation of Fst. The provided file must contain a list of individuals (one individual per line) from the VCF file that correspond to one popula-
tion. This option can be used multiple times to calculate Fst for more than two populations. These files will also be included as "--keep" options. By default, calculations are done on a per-site basis. The output file has the suffix ".weir.fst".
--fst-window-size <integer>
--fst-window-step <integer>
These options can be used with "--weir-fst-pop" to do the Fst calculations on a windowed basis instead of a per-site basis. These arguments specify the desired window size and the desired step size between windows.
OUTPUT OTHER STATISTICS
--het
Calculates a measure of heterozygosity on a per-individual basis. Specfically, the inbreeding coefficient, F, is estimated for each individual using a method of moments. The resulting file has the suffix ".het".
--hardy
Reports a p-value for each site from a Hardy-Weinberg Equilibrium test (as defined by Wigginton, Cutler and Abecasis (2005)). The resulting file (with suffix ".hwe") also contains the Observed numbers of Homozygotes and Heterozygotes and the corresponding
Expected numbers under HWE.
--TajimaD <integer>
Outputs Tajima's D statistic in bins with size of the specified number. The output file has the suffix ".Tajima.D".
--indv-freq-burden
This option calculates the number of variants within each individual of a specific frequency. The resulting file has the suffix ".ifreqburden".
--LROH
This option will identify and output Long Runs of Homozygosity. The output file has the suffix ".LROH". This function is experimental, and will use a lot of memory if applied to large datasets.
--relatedness
This option is used to calculate and output a relatedness statistic based on the method of Yang et al, Nature Genetics 2010 (doi:10.1038/ng.608). Specifically, calculate the unadjusted Ajk statistic. Expectation of Ajk is zero for individuals within a popu-
lations, and one for an individual with themselves. The output file has the suffix ".relatedness".
--relatedness2
This option is used to calculate and output a relatedness statistic based on the method of Manichaikul et al., BIOINFORMATICS 2010 (doi:10.1093/bioinformatics/btq559). The output file has the suffix ".relatedness2".
--site-quality
Generates a file containing the per-site SNP quality, as found in the QUAL column of the VCF file. This file has the suffix ".lqual".
--missing-indv
Generates a file reporting the missingness on a per-individual basis. The file has the suffix ".imiss".
--missing-site
Generates a file reporting the missingness on a per-site basis. The file has the suffix ".lmiss".
--SNPdensity <integer>
Calculates the number and density of SNPs in bins of size defined by this option. The resulting output file has the suffix ".snpden".
--kept-sites
Creates a file listing all sites that have been kept after filtering. The file has the suffix ".kept.sites".
--removed-sites
Creates a file listing all sites that have been removed after filtering. The file has the suffix ".removed.sites".
--singletons
This option will generate a file detailing the location of singletons, and the individual they occur in. The file reports both true singletons, and private doubletons (i.e. SNPs where the minor allele only occurs in a single individual and that individual
is homozygotic for that allele). The output file has the suffix ".singletons".
--hist-indel-len
This option will generate a histogram file of the length of all indels (including SNPs). It shows both the count and the percentage of all indels for indel lengths that occur at least once in the input file. SNPs are considered indels with length zero. The
output file has the suffix ".indel.hist".
--hapcount <BED file>
This option will output the number of unique haplotypes within user specified bins, as defined by the BED file. The output file has the suffix ".hapcount".
--mendel <PED file>
This option is use to report mendel errors identified in trios. The command requires a PLINK-style PED file, with the first four columns specifying a family ID, the child ID, the father ID, and the mother ID. The output of this command has the suffix
".mendel".
--extract-FORMAT-info <string>
Extract information from the genotype fields in the VCF file relating to a specfied FORMAT identifier. The resulting output file has the suffix ".<FORMAT_ID>.FORMAT". For example, the following command would extract the all of the GT (i.e. Genotype)
entries:
vcftools --vcf file1.vcf --extract-FORMAT-info GT
--get-INFO <string>
This option is used to extract information from the INFO field in the VCF file. The <string> argument specifies the INFO tag to be extracted, and the option can be used multiple times in order to extract multiple INFO entries. The resulting file, with suf-
fix ".INFO", contains the required INFO information in a tab-separated table. For example, to extract the NS and DB flags, one would use the command:
vcftools --vcf file1.vcf --get-INFO NS --get-INFO DB
OUTPUT VCF FORMAT
--recode
--recode-bcf
These options are used to generate a new file in either VCF or BCF from the input VCF or BCF file after applying the filtering options specified by the user. The output file has the suffix ".recode.vcf" or ".recode.bcf". By default, the INFO fields are
removed from the output file, as the INFO values may be invalidated by the recoding (e.g. the total depth may need to be recalculated if individuals are removed). This behavior may be overriden by the following options. By default, BCF files are written out
as BGZF compressed files.
--recode-INFO <string>
--recode-INFO-all
These options can be used with the above recode options to define an INFO key name to keep in the output file. This option can be used multiple times to keep more of the INFO fields. The second option is used to keep all INFO values in the original file.
--contigs <string>
This option can be used in conjuction with the --recode-bcf when the input file does not have any contig declarations. This option expects a file name with one contig header per line. These lines are included in the output file.
OUTPUT OTHER FORMATS
--012
This option outputs the genotypes as a large matrix. Three files are produced. The first, with suffix ".012", contains the genotypes of each individual on a separate line. Genotypes are represented as 0, 1 and 2, where the number represent that number of
non-reference alleles. Missing genotypes are represented by -1. The second file, with suffix ".012.indv" details the individuals included in the main file. The third file, with suffix ".012.pos" details the site locations included in the main file.
--IMPUTE
This option outputs phased haplotypes in IMPUTE reference-panel format. As IMPUTE requires phased data, using this option also implies --phased. Unphased individuals and genotypes are therefore excluded. Only bi-allelic sites are included in the output.
Using this option generates three files. The IMPUTE haplotype file has the suffix ".impute.hap", and the IMPUTE legend file has the suffix ".impute.hap.legend". The third file, with suffix ".impute.hap.indv", details the individuals included in the haplo-
type file, although this file is not needed by IMPUTE.
--ldhat
--ldhelmet
--ldhat-geno
These options output data in LDhat/LDhelmet format. This option requires the "--chr" filter option to also be used. The two first options output phased data only, and therefore also implies "--phased" be used, leading to unphased individuals and genotypes
being excluded. For LDhelmet, only snps will be considered, and therefore it implies "--remove-indels". The second option treats all of the data as unphased, and therefore outputs LDhat files in genotype/unphased format. Two output files are generated with
the suffixes ".ldhat.sites" and ".ldhat.locs", which correspond to the LDhat "sites" and "locs" input files respectively; for LDhelmet, the two files generated have the suffixes ".ldhelmet.snps" and ".ldhelmet.pos", which corresponds to the "SNPs" and
"positions" files.
--BEAGLE-GL
--BEAGLE-PL
These options output genotype likelihood information for input into the BEAGLE program. The VCF file is required to contain FORMAT fields with "GL" or "PL" tags, which can generally be output by SNP callers such as the GATK. Use of this option requires a
chromosome to be specified via the "--chr" option. The resulting output file has the suffix ".BEAGLE.GL" or ".BEAGLE.PL" and contains genotype likelihoods for biallelic sites. This file is suitable for input into BEAGLE via the "like=" argument.
--plink
--plink-tped
--relatedness
This option is used to calculate and output a relatedness statistic based on the method of Yang et al, Nature Genetics 2010 (doi:10.1038/ng.608). Specifically, calculate the unadjusted Ajk statistic. Expectation of Ajk is zero for individuals within a popu-
lations, and one for an individual with themselves. The output file has the suffix ".relatedness".
--relatedness2
This option is used to calculate and output a relatedness statistic based on the method of Manichaikul et al., BIOINFORMATICS 2010 (doi:10.1093/bioinformatics/btq559). The output file has the suffix ".relatedness2".
--site-quality
Generates a file containing the per-site SNP quality, as found in the QUAL column of the VCF file. This file has the suffix ".lqual".
--missing-indv
Generates a file reporting the missingness on a per-individual basis. The file has the suffix ".imiss".
--missing-site
Generates a file reporting the missingness on a per-site basis. The file has the suffix ".lmiss".
--SNPdensity <integer>
Calculates the number and density of SNPs in bins of size defined by this option. The resulting output file has the suffix ".snpden".
--kept-sites
Creates a file listing all sites that have been kept after filtering. The file has the suffix ".kept.sites".
--removed-sites
Creates a file listing all sites that have been removed after filtering. The file has the suffix ".removed.sites".
--singletons
This option will generate a file detailing the location of singletons, and the individual they occur in. The file reports both true singletons, and private doubletons (i.e. SNPs where the minor allele only occurs in a single individual and that individual
is homozygotic for that allele). The output file has the suffix ".singletons".
--hist-indel-len
This option will generate a histogram file of the length of all indels (including SNPs). It shows both the count and the percentage of all indels for indel lengths that occur at least once in the input file. SNPs are considered indels with length zero. The
output file has the suffix ".indel.hist".
--hapcount <BED file>
This option will output the number of unique haplotypes within user specified bins, as defined by the BED file. The output file has the suffix ".hapcount".
--mendel <PED file>
This option is use to report mendel errors identified in trios. The command requires a PLINK-style PED file, with the first four columns specifying a family ID, the child ID, the father ID, and the mother ID. The output of this command has the suffix
".mendel".
--extract-FORMAT-info <string>
Extract information from the genotype fields in the VCF file relating to a specfied FORMAT identifier. The resulting output file has the suffix ".<FORMAT_ID>.FORMAT". For example, the following command would extract the all of the GT (i.e. Genotype)
entries:
vcftools --vcf file1.vcf --extract-FORMAT-info GT
--get-INFO <string>
This option is used to extract information from the INFO field in the VCF file. The <string> argument specifies the INFO tag to be extracted, and the option can be used multiple times in order to extract multiple INFO entries. The resulting file, with suf-
fix ".INFO", contains the required INFO information in a tab-separated table. For example, to extract the NS and DB flags, one would use the command:
vcftools --vcf file1.vcf --get-INFO NS --get-INFO DB
OUTPUT VCF FORMAT
--recode
--recode-bcf
These options are used to generate a new file in either VCF or BCF from the input VCF or BCF file after applying the filtering options specified by the user. The output file has the suffix ".recode.vcf" or ".recode.bcf". By default, the INFO fields are
removed from the output file, as the INFO values may be invalidated by the recoding (e.g. the total depth may need to be recalculated if individuals are removed). This behavior may be overriden by the following options. By default, BCF files are written out
as BGZF compressed files.
--recode-INFO <string>
--recode-INFO-all
These options can be used with the above recode options to define an INFO key name to keep in the output file. This option can be used multiple times to keep more of the INFO fields. The second option is used to keep all INFO values in the original file.
--contigs <string>
This option can be used in conjuction with the --recode-bcf when the input file does not have any contig declarations. This option expects a file name with one contig header per line. These lines are included in the output file.
OUTPUT OTHER FORMATS
--012
This option outputs the genotypes as a large matrix. Three files are produced. The first, with suffix ".012", contains the genotypes of each individual on a separate line. Genotypes are represented as 0, 1 and 2, where the number represent that number of
non-reference alleles. Missing genotypes are represented by -1. The second file, with suffix ".012.indv" details the individuals included in the main file. The third file, with suffix ".012.pos" details the site locations included in the main file.
--IMPUTE
This option outputs phased haplotypes in IMPUTE reference-panel format. As IMPUTE requires phased data, using this option also implies --phased. Unphased individuals and genotypes are therefore excluded. Only bi-allelic sites are included in the output.
Using this option generates three files. The IMPUTE haplotype file has the suffix ".impute.hap", and the IMPUTE legend file has the suffix ".impute.hap.legend". The third file, with suffix ".impute.hap.indv", details the individuals included in the haplo-
type file, although this file is not needed by IMPUTE.
--ldhat
--ldhelmet
--ldhat-geno
These options output data in LDhat/LDhelmet format. This option requires the "--chr" filter option to also be used. The two first options output phased data only, and therefore also implies "--phased" be used, leading to unphased individuals and genotypes
being excluded. For LDhelmet, only snps will be considered, and therefore it implies "--remove-indels". The second option treats all of the data as unphased, and therefore outputs LDhat files in genotype/unphased format. Two output files are generated with
the suffixes ".ldhat.sites" and ".ldhat.locs", which correspond to the LDhat "sites" and "locs" input files respectively; for LDhelmet, the two files generated have the suffixes ".ldhelmet.snps" and ".ldhelmet.pos", which corresponds to the "SNPs" and
"positions" files.
--BEAGLE-GL
--BEAGLE-PL
These options output genotype likelihood information for input into the BEAGLE program. The VCF file is required to contain FORMAT fields with "GL" or "PL" tags, which can generally be output by SNP callers such as the GATK. Use of this option requires a
chromosome to be specified via the "--chr" option. The resulting output file has the suffix ".BEAGLE.GL" or ".BEAGLE.PL" and contains genotype likelihoods for biallelic sites. This file is suitable for input into BEAGLE via the "like=" argument.
--plink
--plink-tped
--chrom-map
These options output the genotype data in PLINK PED format. With the first option, two files are generated, with suffixes ".ped" and ".map". Note that only bi-allelic loci will be output. Further details of these files can be found in the PLINK documenta-
tion.
Note: The first option can be very slow on large datasets. Using the --chr option to divide up the dataset is advised, or alternatively use the --plink-tped option which outputs the files in the PLINK transposed format with suffixes ".tped" and ".tfam".
For usage with variant sites in species other than humans, the --chrom-map option may be used to specify a file name that has a tab-delimited mapping of chromosome name to a desired integer value with one line per chromosome. This file must contain a map-
ping for every chromosome value found in the file.
COMPARISON OPTIONS
These options are used to compare the original variant file to another variant file and output the results. All of the diff functions require both files to contain the same chromosomes and that the files be sorted in the same order. If one of the files contains
chromosomes that the other file does not, use the --not-chr filter to remove them from the analysis.
DIFF VCF FILE
--diff <filename>
--gzdiff <filename>
--diff-bcf <filename>
These options compare the original input file to this specified VCF, gzipped VCF, or BCF file. These options must be specified with one additional option described below in order to specify what type of comparison is to be performed. See the examples sec-
tion for typical usage.
DIFF OPTIONS
--diff-site
Outputs the sites that are common / unique to each file. The output file has the suffix ".diff.sites_in_files".
--diff-indv
Outputs the individuals that are common / unique to each file. The output file has the suffix ".diff.indv_in_files".
--diff-site-discordance
This option calculates discordance on a site by site basis. The resulting output file has the suffix ".diff.sites".
--diff-indv-discordance
This option calculates discordance on a per-individual basis. The resulting output file has the suffix ".diff.indv".
--diff-indv-map <filename>
This option allows the user to specify a mapping of individual IDs in the second file to those in the first file. The program expects the file to contain a tab-delimited line containing an individual's name in file one followed by that same individual's
name in file two with one mapping per line.
--diff-discordance-matrix
This option calculates a discordance matrix. This option only works with bi-allelic loci with matching alleles that are present in both files. The resulting output file has the suffix ".diff.discordance.matrix".
--diff-switch-error
This option calculates phasing errors (specifically "switch errors"). This option creates an output file describing switch errors found between sites, with suffix ".diff.switch".
AUTHORS
Adam Auton (adam.auton@einstein.yu.edu)
Anthony Marcketta (anthony.marcketta@einstein.yu.edu)
実行方法
gzip圧縮したvcfと非圧縮vcf(一部コマンドのみ)を両方扱うことができる。
#もし圧縮するなら (bgzipがないなら”conda install -c bioconda tabix”)
bgzip input.vcf
tabix -p vcf input.vcf.gz
#解凍
bgzip -d input.vcf.gz
#解凍(元のファイルは残す)
bgzip -dc input.vcf.gz > output.vcf
vcf format4.3を無理やり4.2に見せかける(非推奨)。#biostarのスレッドより(link) 。
#v4.3 => 4.2
sed 's/^##fileformat=VCFv4.3/##fileformat=VCFv4.2/' in.vcf > out.vcf
特定のchrだけ出力する。非圧縮vcfは--vcfで、gzip圧縮したvcfは--gzvcf で指定する。
#output only chr1 call
vcftools --vcf input_file.vcf --recode --out output --chr 1
#output only chr1 call, and includes all INFO fields
vcftools --vcf input_file.vcf --recode --out output --chr 1 --recode-INFO-all
#remove chr1 and chr2 call
vcftools --vcf input_file.vcf --recode --out output --not-chr 1 --not-chr 2
#output only chr21:1-500000 call
vcftools --vcf input_file.vcf --recode --out output --chr 21 --from-bp 1 --to-bp 500000
- --recode, --recode-bcf These options are used to generate a new file in either VCF or BCF from the input VCF or BCF file after applying the filtering options specified by the user. The output file has the suffix ".recode.vcf" or ".recode.bcf". By default, the INFO fields are removed from the output file, as the INFO values may be invalidated by the recoding (e.g. the total depth may need to be recalculated if individuals are removed). This behavior may be overriden by the following options. By default, BCF files are written out as BGZF compressed files.
- --recode-INFO <string>, --recode-INFO-all These options can be used with the above recode options to define an INFO key name to keep in the output file. This option can be used multiple times to keep more of the INFO fields. The second option is used to keep all INFO values in the original file.
logファイルとvcfファイルが出力される。logファイルには使用したパラメータや残ったコールの数が記録される。

染色体名が分からなければVCFの先頭かfasta.faiを見る。 1番染色体は"chr1"

bcfに変換して出力する。(もちろん他の処理と組み合わせる事が可能)。
vcftools --vcf input.vcf --recode-bcf --recode-INFO-all --out output.bcf
- --recode-bcf These options are used to generate a new file in either VCF or BCF from the input VCF or BCF file after applying the filtering options specified by the user. The output file has the suffix ".recode.vcf" or ".recode.bcf". By default, the INFO fields are
indelを除く。またはindellのみ。非圧縮vcfは--vcfで、gzip圧縮したvcfは--gzvcf で指定する。
#remove indel
vcftools --vcf input_file.vcf --remove-indels --recode --recode-INFO-all --out SNPs
#keep only indel
vcftools --vcf input_file.vcf --keep-only-indels --recode --recode-INFO-all --out indels
- --keep-only-indels, --remove-indels Include or exclude sites that contain an indel. For these options "indel" means any variant that alters the length of the REF allele.
共通 / 固有のバリアントを出力。非圧縮vcfは--vcfで、gzip圧縮したvcfは--gzvcf で指定する。
vcftools --gzvcf input_file1.vcf.gz --gzdiff input_file2.vcf.gz --diff-site --out output
- --diff-site Outputs the sites that are common / unique to each file. The output file has the suffix ".diff.sites_in_files".
- --diff-indv Outputs the individuals that are common / unique to each file. The output file has the suffix ".diff.indv_in_files".
- --diff-site-discordance This option calculates discordance on a site by site basis. The resulting output file has the suffix ".diff.sites".
- --diff-indv-discordance This option calculates discordance on a per-individual basis. The resulting output file has the suffix ".diff.indv".
- --diff-indv-map <filename> This option allows the user to specify a mapping of individual IDs in the second file to those in the first file. The program expects the file to contain a tab-delimited line containing an individual's name in file one followed by that same individual's name in file two with one mapping per line.
- --diff-discordance-matrix This option calculates a discordance matrix. This option only works with bi-allelic loci with matching alleles that are present in both files. The resulting output file has the suffix ".diff.discordance.matrix".
- --diff-switch-error This option calculates phasing errors (specifically "switch errors"). This option creates an output file describing switch errors found between sites, with suffix ".diff.switch". would use the command:
Manichaikul et al. (doi:10.1093/bioinformatics/btq559)の方法に基づいて、関連統計量を出力。
vcftools --vcf input_file.vcf --relatedness2
出力のsuffixは".relatedness2"。
Transition / Transversion比を計算。
vcftools --vcf input_file.vcf --TsTv-by-count
vcftools --vcf input_file.vcf --TsTv-by-qual
vcftools --vcf input_file.vcf --TsTv-summary
-
--TsTv-summary Calculates a simple summary of all Transitions and Transversions. The output file has the suffix ".TsTv.summary".
-
--TsTv-by-count output file has the suffix ".TsTv.count".
-
--TsTv-by-qual Calculates the Transition / Transversion ratio as a function of SNP quality threshold. Only uses bi-allelic SNPs. The resulting output file has the suffix ".TsTv.qual".
2つのVCFファイルを比較して、どのサイトと個人が共有されているかを判断する。2つ目のVCFは --diff, --gzdiff, --diff-bcf を使って指定する。
vcftools --vcf input_data.vcf --diff other_data.vcf --out compare
- --diff <filename>, --gzdiff <filename>, --diff-bcf <filename> These options compare the original input file to this specified VCF, gzipped VCF, or BCF file. These options must be specified with one additional option described below in order to specify what type of comparison is to be performed. See the examples section for typical usage.
他にも複数コマンドがあります。
以下は ソースからビルドした場合のみ(condaで導入した場合は認識されなかった)。
2つ以上のVCFをマージする。
vcf-merge A.vcf.gz B.vcf.gz C.vcf.gz | bgzip -c > out.vcf.gz
#追記bcftoolsでマージ
bcftools merge --merge all --force-samples \
wt.vcf.gz mt1.vcf.gz mt2.vcf.gz > merged.vcf
ポジションが同じ部位は1行にまとめられる。右端の4列を見ればどうなっているかわかる。
AとBに存在。Cにはない。
GT:GQ:DP:PL:AD 1/1:75:25:1115,75,0:0,25 1/1:60:37:795,60,0:0,37 .
Aだけに存在。
GT:GQ:DP:PL:AD 1/1:99:59:2415,181,0:0,59 . .
SVcallの結果をマージするならSURVIVORが便利です。
SVシミュレーションや、SVのマージ、レポート生成ができる SURVIVOR
2つ以上のVCFをコメント(##)を除き結合する。(ポジションが同じ部位をマージしたりソートしたりはしない)。
vcf-concat A.vcf.gz B.vcf.gz C.vcf.gz | gzip -c > out.vcf.gz
chrとポジションでソートする。
vcf-sort file.vcf.gz
ソートコマンドの sort -k1,1d -k2,2n(1列目ポジションソート+2列目ポジションソート)を実行したのと同じ結果になる。
VCFのバリアント部位を元にレファレンスfastaを改変する。
cat ref.fa | vcf-consensus file.vcf.gz > out.fa
- --sample <name> If not given, all variants are applied
- --haplotype <int> Apply only variants for the given haplotype (1,2)
- --iupac-codes Apply variants in the form of IUPAC ambiguity codes
VCFのフォーマットを3.3から4.0に変える。
zcat file.vcf.gz | vcf-convert -r reference.fa > out.vcf
- --version <string> 4.0, 4.1, 4.2
共通のバリアントや固有のバリアントを検出する。
2つ以上のVCFに共通して存在しているバリアントを抽出。
vcf-isec -n +2 A.vcf.gz B.vcf.gz | bgzip -c > out.vcf.gz
- -n [+-=]<int> Output positions present in this many (=), this many or more (+), or this many or fewer (-) files.
Aに存在して、B、Cには存在しないバリアントを抽出。カラム名が違うというエラー(異なるVCFである)というエラーを防ぐには"-f"オプションをつける。
vcf-isec -c A.vcf.gz B.vcf.gz C.vcf.gz | bgzip -c > out.vcf.gz
- -c Output positions present in the first file but missing from the other files.
- -f Continue even if the script complains about differing columns, VCF versions, etc.
- -p <path> If present, multiple files will be created with all possible isec combinations. (Suitable for Venn Diagram analysis.)
- -r <list|file> Do only the given regions (comma-separated list or one region per line in a file).
- -w <int> In repetitive sequences, the same indel can be called at different positions. Consider records this far apart as matching (be it a SNP or an indel).
MQが30以上の部位を"PASS"させる。(MQリンク)
vcf-annotate -f MinMQ=30 input.vcf > output.vcf
- -f <file|list> Apply filters, list is in the format flt1=value/flt2/flt3=value/etc. If argument to -f is a file, user-defined filters be applied. See User Defined Filters below.
- -H Remove lines with FILTER anything else than PASS or "."
- -r <list> Comma-separated list of tags to be removed (e.g. ID,INFO/DP,FORMAT/DP,FILTER).
出力を確認すると、MQが30以上のものがフィルターをPASSしている。

デプス、アレルのデプス、マッピングクオリティなどのフィルターをかける。
vcf-annotate -f +/-a/c=3,10/d=10/-D/MinMQ=40/ input.vcf > output.vcf
以下のようなフィルターがある。

上ではMinMQと書いているが、qと書いてもOK。PASSしなかった部位はその名前がつく。MinDPが通らなかったらFilterのカラムにMinDPと付く。
追記
VCFのバージョンなどでフィルタリングがうまく機能しない例があるようです。その場合はvcflib(bioconda)(インストールは"conda install -c bioconda vcflib")のvcffilterのフィルタリングを試してみてください。例えば、 DP>10、MQ>30、QD>20でフィルタリング。カバレッジの薄い領域、レアバリアントの大半は除去されます。docker imageもあります(pull数が多いイメージ例: docker pull shollizeck/vcflib:latest)。
(注;例えばMQタグがないVCFもあるので、その時はフィルタリングでエラーになる。grepで該当するタグがVCFファイル中に存在するか確認しておくこと)
#ここではvcfilterのバージョン差による違いを避けるためにdockerイメージを使う
docker run -itv $PWD:/data --rm shollizeck/vcflib:latest
vcffilter -h
cd /data/
#クオリティフィルタリング
vcffilter -f "DP > 10 & MQ > 30 & QD > 20" input.vcf > filtered.vcf
#cannot comapreのエラーがでるなら、そのtagがVCFに存在しない可能性がある。grepで確認してください => grep "MQ" in.vcf
#FreebayesではMQの代わりにMQMが使える
vcffilter -f "DP > 10 & MQM > 30" Freebayes.vcf > Freebayes_filtered.vcf
#それからhomozygousのコールだけ抽出。
cat filtered.vcf | vcffilter -g "GT = 1/1" | vcffixup - | vcffilter -f "AC > 0" > homozygous.vcf
#圧縮
bgzip results.vcf
tabix -p vcf results.vcf.gz
IGVでみる場合、preference => variantのタブでバリアントのカラーの調整ができます。

vcf-mergeなどでマージしたvcfをIGVに読み込ませると、mergeしたvcf.gzにぶら下がってどのツール由来のコールかも表示されます。

VCFを比較して固有のバリアントと共通のバリアントの数を出力する(ベン図を書くときに使える)。
vcf-compare A.vcf.gz B.vcf.gz C.vcf.gz > output
- -a Ignore lines where FILTER column is anything else than PASS or '.'
- -c <list|file> Same as -r, left for backward compatibility. Please do not use as it will be dropped in the future.
- -d Debugging information. Giving the option multiple times increases verbosity
- --cmp-genotypes Compare genotypes, not only positions
- -ignore-indels Exclude sites containing indels from genotype comparison
- -R <file> Compare the actual sequence, not just positions. Use with -w to compare indels.
VCF2ファイルを比較すると以下のような出力が得られる。
o$ cat out
# This file was generated by vcf-compare.
# The command line was: vcf-compare(v0.1.14-12-gcdb80b8) HaplotypeCaller.vcf.gz UnifiedGenotyper.vcf.gz
#
#VN 'Venn-Diagram Numbers'. Use `grep ^VN | cut -f 2-` to extract this part.
#VN The columns are:
#VN 1 .. number of sites unique to this particular combination of files
#VN 2- .. combination of files and space-separated number, a fraction of sites in the file
VN 5 UnifiedGenotyper.vcf.gz (7.9%) #VCF1固有
VN 19 HaplotypeCaller.vcf.gz (24.7%) #VCF2固有
VN 58 HaplotypeCaller.vcf.gz (75.3%) UnifiedGenotyper.vcf.gz (92.1%) #共通
#SN Summary Numbers. Use `grep ^SN | cut -f 2-` to extract this part.
SN Number of REF matches: 58
SN Number of ALT matches: 57
SN Number of REF mismatches: 0
SN Number of ALT mismatches: 1
SN Number of samples in GT comparison: 0
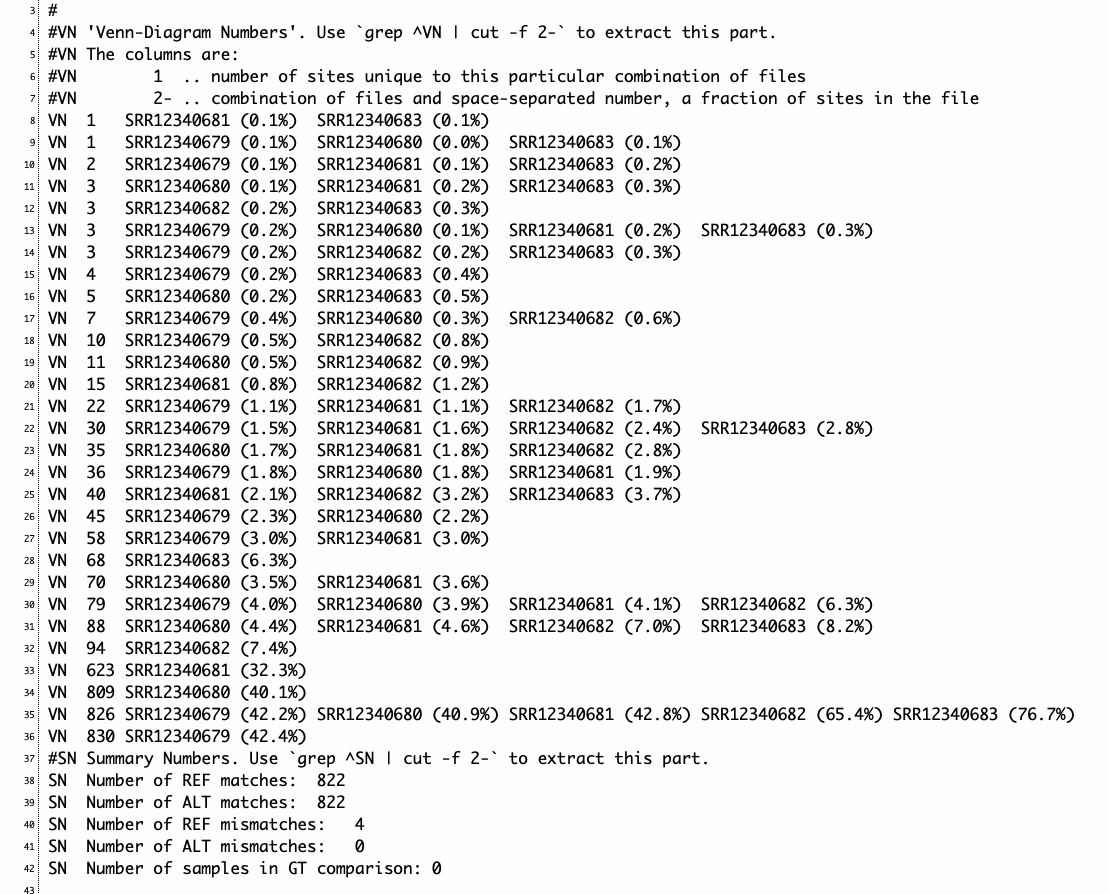
5個のVCFを比較(それぞれのユニークなサイトが数の昇順で表示される。例えば、8行目はSRR12340681とSRR12340683だけ共通して見つかるバリアントが1つだけあることを示している)
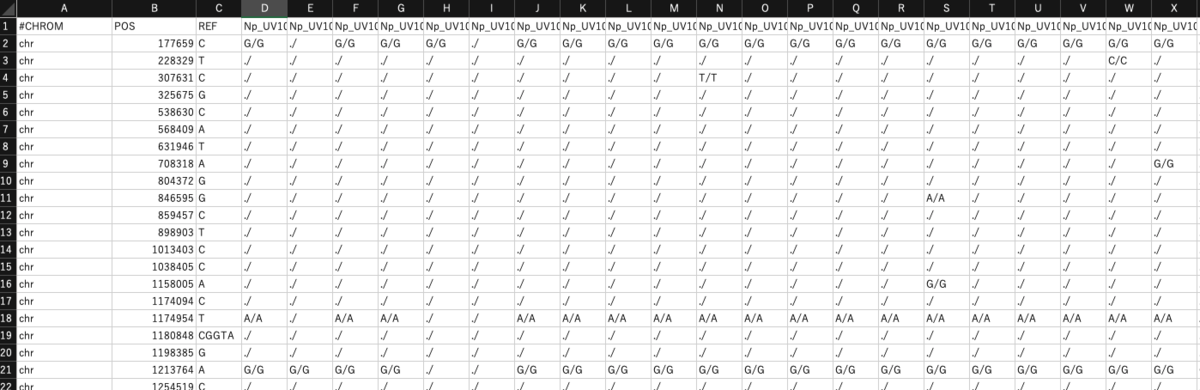
VCFをシンプルなタブ形式に変換する。
zcat file.vcf.gz | vcf-to-tab > out.tab
#REFと同じサンプルは”./”になっているが、vcfが巨大だと消したい時がある。sedで消す。
sed s/'\.\/'/''/g out.tab > remove.tab
以下のような出力が得られる。
user$ head -6 out.tab
#CHROM POS REF Z
chr 831647 C T/T
chr 943495 G A/A
chr 1012958 G T/T
chr 1114645 T TTTAATTAGGGTGATAAATTCG/TTTAATTAGGGTGATAAATTCG
chr 1204616 G A/A
複数サンプル含まれるVCFを変換した場合
SNPs、indelの要約統計。
#VCFの中のサンプルNA00001だけ分析
vcf-stats file.vcf.gz -f SAMPLE/NA00001/DP=1:200 -p out/
#VCFの中の全サンプルを分析
vcf-stats file.vcf.gz -f FORMAT/DP=10:200 -p out/
#bcftoolsを使うなら(VCFの中の全サンプル)
bcftools stats -F ref.fasta -s mpileup.vcf.gz > mpileup.vcf.gz.stats
#create report pd
plot-vcfstats -p report/ mpileup.vcf.gz.stats
vcf-contrast
グループ間の違いを見つける。
#サンプルA,Bのいずれかが、すべてのC,D,Eと異なるかどうかを調べる。最低20のMAOQ設定。
vcf-compare -f MinMQ=20 A.vcf.gz B.vcf.gz C.vcf.gz > output
fill-an-ac
ANおよびAC INFOフィールドの入力または再計算。
fill-an-ac [OPTIONS] in.vcf > out.vcf
fill-fs
VCFファイルの既知のバリアントをNでマスクし、フランキングシーケンス(INFO/FSタグ)をアノテーションする。プライマーの設計に便利。
#VCFファイルのバリアントをNでマスクし、ベッドファイルのリージョンを小文字にする。
fill-fs file.vcf -v mask.vcf -m lc -b mask.bed
fill-ref-md5
不足しているリファレンス情報を埋め、MD5をVCFのヘッダーに配列する。
fill-ref-md5 -i AS:NCBIM37,SP:"Mus\ Musculus" -r NCBIM37_um.fa -d NCBIM37_um.fa.dict in.vcf.gz out.vcf.gz
vcf-indel-stats
in-frame比率の計算。
vcf-indel-stats [OPTIONS] < in.vcf > out.txt
vcf-subset
VCFファイルからいくつかの列を削除する。
cat in.vcf | vcf-subset -r -t indels -e -c SAMPLE1 > out.vcf
vcf-query
VCFファイルをユーザーが定義したフォーマットに変換する強力なコマンド。ポジション、カラム、フィールドのサブセットの検索をサポートする。
vcf-query file.vcf.gz 1:1000-2000 -c NA001,NA002,NA003
vcf-shuffle-cols
VCFの列を並び替える。
vcf-shuffle-cols [OPTIONS] -t template.vcf.gz file.vcf.gz > out.vcf
vcf-annotate The script adds or removes filters and custom annotations to VCF files. To add custom annotations to VCF files, create TAB delimited file with annotations such as
vcf-fix-newlines Fixes diploid vs haploid genotypes on sex chromosomes, including the pseudoautosomal regions.
vcf-fix-ploidy Fixes diploid vs haploid genotypes on sex chromosomes, including the pseudoautosomal regions.
vcf-phased-join Concatenates multiple overlapping VCFs preserving phasing.
追記
例えばvarscan2でバリアントコール(SNPsとsmall indels)し、VCFtoolsでSNPs と indelを分けて出力する。
#pileup
sambamba mpileup input.bam -t 12 --samtools -f ref.fa > pileup
#varscanのmpileup2vcfでバリアントコール
varscan mpileup2vcf mpileup --output-vcf 1 --variants 1 \
--min-coverage 8 --min-reads2 4 --min-avg-qual 15 --min-var-freq 0.01 \
--min-freq-for-hom 0.75 --p-value 99e-02 \
> varscan.vcf
#フィルタリング1 SNV
vcftools --vcf varscan.vcf --remove-indels --recode --recode-INFO-all --out varscan_SNV.vcf
#フィルタリング2 indel
vcftools --vcf varscan.vcf --keep-only-indels --recode --recode-INFO-all --out varscan_indel.vcf
引用
The variant call format and VCFtools
Petr Danecek,1,† Adam Auton,2,† Goncalo Abecasis,3 Cornelis A. Albers,1 Eric Banks,4 Mark A. DePristo,4 Robert E. Handsaker,4 Gerton Lunter,2 Gabor T. Marth,5 Stephen T. Sherry,6 Gilean McVean,2,7 Richard Durbin,1,* and 1000 Genomes Project Analysis Groupcorresponding author
Bioinformatics. 2011 Aug 1; 27(15): 2156–2158.
BIostars 質問 VCFのマージ
https://www.biostars.org/p/49730/
追記
変異が数十万以上あって動作が緩慢なら、最初にcyvcf2で不要な情報を除外してから使ってください。cyvcf2は2017年に発表された高速なVCFのパーサです。