2024/05/22 追記、誤字修正、コメント追加
2024/08/22 追記
2025/03/04 追記
メタゲノムの分類学的な分類器は、DNA配列かアミノ酸(AA)配列のどちらかを解析する。しかし、Metabuli (https://metabuli.steineggerlab.com)は、DNAとAAの両方を共同で解析し、感度の高い相同性検出のためにAAの保存性を活用し、近縁の分類群を特異的に区別するためにDNA変異を活用する。Critical Assessment of Metagenome Interpretation 2 (CAMI2) plant-associated datasetにおいて、Metabuliは最先端のDNAベースおよびAAベースの分類器の分類をそれぞれ99%および98%カバーした。
Metabuli introduces a new compact k-mer encoding, metamer, which stores AA and their codons. For sensitivity, metamers are first compared on AA level for exact matches, and subsequently, for high specificity, are compared on the DNA level to distinguish close matches. 4/n pic.twitter.com/oM7OFnncc4
— Jaebeom Kim (@jbeom_kim) 2024年5月20日
Metabuli is freely available through github, compiled binary, and Biocodna. Try Metabuli with your data using pre-built databases of GTDB and prokaryotic and viral RefSeq. 2/n
— Jaebeom Kim (@jbeom_kim) 2024年5月20日
🌐https://t.co/bjEjqBTlaN
Metabuli(분리), our metagenomic read classifier, is now published @naturemethods. By combining DNA & AA information it recovers 99% and 98% assignments of DNA or AA classifiers. Great work by my student @jbeom_kim 1/n
— Martin Steinegger 🇺🇦 (@thesteinegger) 2024年5月20日
💾https://t.co/QkZx5CJvv6
📄https://t.co/KyfS2TjjLH
レポジトリより
Metabuliは、DNA配列とアミノ酸(AA)配列の両方を共同で解析するメタゲノム分類器である。DNAベースの分類子は、近縁の分類群を区別するための点変異を識別し、特異的な分類を行うことができる。AAベースの分類器は、AA配列の高い保存性を利用して、クエリ配列と参照配列間の相同性を検出する感度が高い。Metabuliは、メタゲノムサンプルの特異的かつ高感度な特性解析を可能にするため、新規のk-mer構造であるmetamerを用いて、両方の配列タイプの情報を組み合わせている。さらに、ハードディスクに収まる限り、あらゆるサイズのデータベースに対してリードを分類することができる。
インストール
ubuntu22.04でテストした。また、 M1 macstudioにもダウンロードして動作確認した。
#conda
mamba install -c conda-forge -c bioconda metabuli -y
# Linux AVX2 build
# check using: cat /proc/cpuinfo | grep avx2)
wget https://mmseqs.com/metabuli/metabuli-linux-avx2.tar.gz
tar xvzf metabuli-linux-avx2.tar.gz
cd metabuli/bin/
export PATH=$(pwd):$PATH
# Linux SSE2 build
wget https://mmseqs.com/metabuli/metabuli-linux-sse2.tar.gz
tar xvzf metabuli-linux-sse2.tar.gz
cd metabuli/bin/
export PATH=$(pwd):$PATH
# MacOS (works on Apple Silicon and Intel Macs)
curl -o metabuli-osx-universal.tar.gz https://mmseqs.com/metabuli/metabuli-osx-universal.tar.gz
tar xvzf metabuli-osx-universal.tar.gz
cd metabuli/bin/
export PATH=$(pwd):$PATH
#comple from source
git clone https://github.com/steineggerlab/Metabuli.git
cd Metabuli
mkdir build && cd build
cmake -DCMAKE_BUILD_TYPE=Release ..
make -j 16
> metabuli -h
Metabuli is a taxonomical classifier that jointly analyzes amino acid and DNA sequences.
metabuli Version: 2923f1cca65360d0d295a0b0b50521bef30e66c3
© Jaebeom Kim <jbeom0731@gmail.com>
usage: metabuli <command> [<args>]
Main workflows for database input/output
classify Assigning taxonomy label to query reads
Input database creation
databases List and download databases
build Build database based on the list of FASTA files.
add-to-library It bins sequences into files according to their species.
Format conversion for downstream processing
binning2report It generates Kraken style report file from binning results
Utility modules to manipulate DBs
database-report It generates a report of taxa in a database.
> ./metabuli classify -h
> ./metabuli classify -h
usage: metabuli classify <i:query file(s)> <i:database directory> <o:output directory> <job ID> [options]
By Jaebeom Kim <jbeom0731@gmail.com>
options: prefilter:
--mask INT Mask sequences in k-mer stage: 0: w/o low complexity masking, 1: with low complexity masking [0]
--mask-prob FLOAT Mask sequences is probablity is above threshold [0.900]
misc:
--seq-mode INT Single-end: 1, Paired-end: 2, Long read: 3 [2]
--min-score FLOAT Min. sequence similarity score (0.0-1.0) [0.000]
--min-cov FLOAT Min. query coverage (0.0-1.0) [0.000]
--min-cons-cnt INT Min. number of consecutive matches for prokaryote/virus classification [4]
--min-cons-cnt-euk INT Min. number of consecutive matches for eukaryote classification [9]
--min-sp-score FLOAT Min. score for species- or lower-level classification. [0.000]
--hamming-margin INT It allows extra Hamming distance than the minimum distance. [0]
--taxonomy-path STR Directory where the taxonomy dump files are stored
--max-ram INT RAM usage in GiB [128]
--match-per-kmer INT Num. of matches per query k-mer. Larger values assign more memory for storing k-mer matches. [4]
--accession-level INT Build or search a database for accession-level classification [0]
--tie-ratio FLOAT Best * --tie-ratio is considered as a tie [0.950]
common:
--threads INT Number of CPU-cores used (all by default) [128]
references:
> metabuli databases -h
usage: metabuli databases <i:Name> <o:OUTDIR> <tmpDir> [options]
By Milot Mirdita <milot@mirdita.de>
Name Type Taxonomy Url
- RefSeq Index yes https://www.ncbi.nlm.nih.gov/refseq/
NCBI RefSeq (Complete Genome/Chromosome level assemblies, prokaryote & virus) and a human genome (T2T-CHM13v2.0)
Cite: O'Leary et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. (2016)
- GTDB Index yes https://gtdb.ecogenomic.org/
GTDB 214.1 (Complete/Chromosome level only, CheckM completeness > 90 and contamination < 5) and a human genome (T2T-CHM13v2.0)
Cite: Donovan et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. (2022)
- RefSeq_virus Index yes https://www.ncbi.nlm.nih.gov/refseq/
NCBI RefSeq release 223 virus genomes and a human genome (T2T-CHM13v2.0)
Cite: O'Leary et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. (2016)
- RefSeq_release Index yes https://www.ncbi.nlm.nih.gov/refseq/
NCBI release 217 (Prokaryote & Virus) and a human genome (GRCh38.p14)
Cite: O'Leary et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. (2016)
options:
--tsv BOOL Return output in TSV format [0]
--force-reuse BOOL Reuse tmp filse in tmp/latest folder ignoring parameters and version changes [0]
--remove-tmp-files BOOL Delete temporary files [0]
--compressed INT Write compressed output [0]
--threads INT Number of CPU-cores used (all by default) [128]
-v INT Verbosity level: 0: quiet, 1: +errors, 2: +warnings, 3: +info [3]
references:
> metabuli build -h
usage: metabuli build <DB dir> <FASTA list> <Accesssion2taxid> [options]
By Jaebeom Kim <jbeom0731@gmail.com>
options: prefilter:
--mask-prob FLOAT Mask sequences is probablity is above threshold [0.900]
--mask INT Mask sequences in k-mer stage: 0: w/o low complexity masking, 1: with low complexity masking [1]
misc:
--reduced-aa INT Set as 0 to use 15 alphabets to encode AAs for sensitivity [0]
--taxonomy-path STR Directory where the taxonomy dump files are stored
--split-num INT A database is divided to N splits (offsets). During classification, unnecessary splits are skipped [4096]
--buffer-size INT Buffer size (the number of k-mers) [1000000000]
--accession-level INT Build or search a database for accession-level classification [0]
--db-name STR Name of the database [1690519666]
--db-date STR Date of the database creation [2024-5-22]
common:
--threads INT Number of CPU-cores used (all by default) [128]
references:
> metabuli add-to-library -h
usage: metabuli add-to-library <I: FASTA list> <I: accession2taxid> <I: DB DIR> [options]
By Jaebeom Kim <jbeom0731@gmail.com>
options: misc:
--assembly BOOL Input is an assembly [0]
--taxonomy-path STR Directory where the taxonomy dump files are stored [DBDIR/taxonomy/]
--library-path STR Path to library where the FASTA files are stored [DBDIR/library/]
references:
データベース
Pre-builtのデータベースが公開されている。refseq_release217+human.tar.gzだとファイルサイズが306.4GBとなっていて、かなり大きい*1。ここではgtdb(2024年5月現在、R214が使用されている)を選択する。しかし株レベルの区別をするにはrefseqの方が適している(要テスト)。ウィルスサーベイならRefSeq_virusとRefSeq_vprocaryotes_virusが選べる。
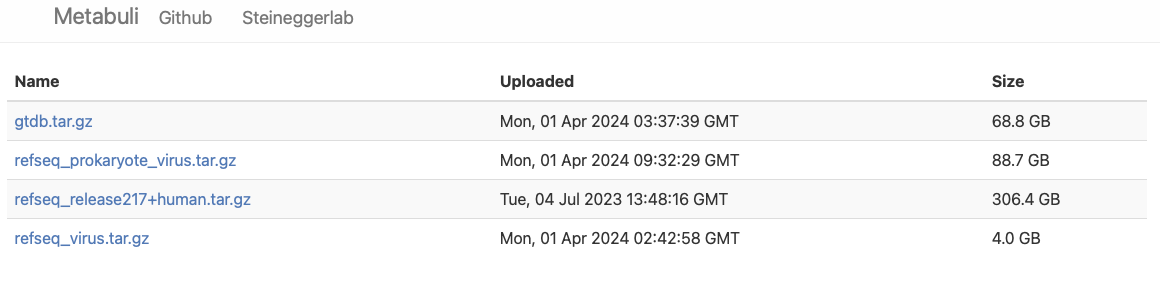 https://metabuli.steineggerlab.workers.dev
https://metabuli.steineggerlab.workers.dev
・ヒトのT2T-CHM13v2.0はRefSeq_releaseを除くすべてのデータベースに収録されている。GRCh38.p14はRefSeq_releaseに収録されている。自前のデータベースをビルドする手順はレポジトリ参照。
Pre-builtのデータベースをダウンロードするコマンドが用意されている。
> metabuli databases

例えばGTDBをDBディレクトリにダウンロードする。tmpは作業ディレクトリ。OUTDIRが出力ディレクトリ名。
metabuli databases GTDB OUTDIR tmp
試した時は2時間くらいでダウンロードできた。
解凍後のファイルサイズは101GB。

(ファイルサイズはGTDBのバージョンによって異なる)
実行方法
ショートもしくはロングのシークエンシングリードのfastaかfastq、DB、出力ディレクトリ、出力されるファイルのprefixとなるjobidの順番で指定する。非圧縮リードとあるが、fastq.gzも認識した(#14)。
# Paired-end
metabuli classify --seq-mode 2 read_1.fna read_2.fna dbdir outdir jobid
# Single-end
metabuli classify --seq-mode 1 read.fna dbdir outdir jobid
# Long-read
metabuli classify --seq-mode 3 read.fna dbdir outdir jobid
- --seq-mode Single-end: 1, Paired-end: 2, Long read: 3 [2]
- --threads The number of threads used (all by default)
- --max-ram The maximum RAM usage. (128 GiB by default)
"--seq-mode"はシークエンシングプラットフォームによって変わるリードの正確性に対応した、精度を決めるパラメータ類のプリセットとなっている(詳細はレポジトリ参照)。
出力例
JobID_classifications.tsv、JobID_report.tsv、JobID_krona.html の3つが出力される。
1,classifications.tsv:各シークエンシングリードについて分類の有無、リードID、分類識別子、有効リード長、DNAレベルの同一性スコア、分類ランクが書かれている。シークエンシングデータサイズが大きいと1GB以上のファイルサイズになる。

2,report.tsv:各分類群に割り当てられたリードの割合が書かれている。

3,krona.html:インタラクティブなkronaのtaxonomy report

論文より
- Metabuliは1,500万イルミナリードを10分で、875,545 PacBio HiFiロングリードを20分で分類し、Kraken2では24秒と76秒だった。注目すべきは、Metabuliはユーザー指定のランダムアクセスメモリー(RAM)制限内で動作し、ストレージにフィットするあらゆるデータベースを検索できることで、8ギガバイトのRAMを搭載したノートブックでもこれらのタスクを完了できる(M1 MacBook airでも動作する)。
- metamerは、DNAとAA情報を共同でエンコードする新規のk-merであり、別々にエンコードする場合に比べ、わずか3分の2のストレージしか必要としない。Metabuliはデータベース作成時に、参照ゲノムのオープンリーディングフレーム(ORF)を予測するためにProdigalを採用し、さらに遺伝子間領域を含むように拡張している。metamerはコドンベースであるため、その保存にはゲノム長の1/3が必要であり、種内の異なるmetamerのみを保存することでさらなる圧縮が達成される。
コメント
1つのコマンドでkrona plotまで作ってくれるのはとても便利ですね。時間があれば、kraken2の翻訳モードとMetabuli間でリアルメタゲノムリードを分類した時の感度と精度の比較をしてみたいのですが、ちょっと時間が無さそうです。感度と言えば、Metabuliが出力するkrona plotには未分類のリード数である"unclassified"の割合もプロットされますので、unclassifiedの割合を見ればMetabuliの感度が十分であったか判断する大まかな指標になるかと思います。一点気になったのは、fastqのサイズが大きいときにランタイムがやや長めだった点です(3990X AVX2有効、全CPU指定でkraken2と比較*2)。ユーザーサイドで高速化するには、読み出しが早いディスクにデータベースを配置する、メインメモリ(RAM)が潤沢に利用できる環境なら(データベースが大きい時に)--max-ramの最大メモリ割り当てを増やす、などが有効かもしれません(要検討)。M1 macbookでも動作するのは非常に魅力的ですね。
レポジトリでは、MetabuliとRefSeq Virus DBを使って、COVID-19患者のRNA-seqリードを分類し、SARS-CoV-2のバリアント(βやγなど)を定量的に評価するフローが紹介されています。興味がある方は確認してみて下さい。
引用
Metabuli: sensitive and specific metagenomic classification via joint analysis of amino acid and DNA
Jaebeom Kim & Martin Steinegger
Nature Methods, Published: 20 May 2024
2025/10/15
Easy and interactive taxonomic profiling with Metabuli App
SunJae Lee, Jaebeom Kim, Milot Mirdita, Cameron L M Gilchrist, Martin Steinegger Author Notes
Bioinformatics, Published: 13 October 2025
関連
・タンパク質を使って高感度にメタゲノムのtaxonomy assignmentを行う kaiju
・メタゲノムのリードの系統アサインメントを行う Centrifuge
*1 kraken2だとさらに巨大なntデータベースも用意されている。
*2 3990Xで、kraken2とMetabuli(AVX2有効)のランタイムを比較した。128スレッド使用時、kraken2は6GBx2のgzip圧縮ペアエンドfastqを3分58秒で分類し、Metabuliは15分13秒で同じリードセットを分類した。条件は、kraken2はNCBI_refseqデータベースを使用して翻訳モード(DNAよりかなり遅くなる)で128スレッド実行した。Metabuliはgtdbデータベースを使用して128スレッド実行した。データベースが異なるので正確な比較にはなっていない。また、Metabuliはkronaレポート作成まで含めてのランタイムなので、色々とフェアな比較が出来ていない点に注意。